Lab-Made? SARS-CoV-2 Genealogy Through the Lens of Gain-of-Function Research

If you hear anyone claim “we know the virus didn’t come from a lab”, don’t buy it — it may well have. Labs around the globe have been creating synthetic viruses like CoV2 for years. And no, its genome would not necessarily contain hallmarks of human manipulation: modern genetic engineering tools permit cutting and pasting genomic fragments without leaving a trace. It can be done quickly, too: it took a Swiss team less than a month to create a synthetic clone of CoV2.
How I Learned to Start Worrying
Oh, come on. Lab-made? Nonsense! Back in January, that was my knee-jerk reaction when ideas that Covid-19 is caused by a laboratory leak had just surfaced. Bioweapon? Well, that is just Flat Earth crazies territory. Thus, whenever I kept hearing anything about non-natural origins of SARS-CoV-2, I brushed it aside under similar sentiments. So what if there is a virology institute in Wuhan? Who knows how many of those are sprinkled throughout China.
At some point, it became necessary to brush such theories aside in a substantiated manner, as their proponents began to back up their theses about the possible artificial nature of the virus with arguments from molecular biology, and when engaging them in debate, I wanted to smash their conspiracy theories with cold, hard scientific facts. Just like that Nature paper (or so I thought).
So it was then, in pursuit of arguments against the virus’s lab-madeness, that I got infected by the virus of doubt. What was the source of my doubts? The fact that the deeper you dive into the research activities of coronavirologists over the past 15–20 years, the more you realize that creating chimeras like CoV2 was commonplace in their labs. And CoV2 is an obvious chimera (though not nesessarily a lab-made one), which is based on the ancestral bat strain RaTG13, in which the receptor binding motif (RBM) in its spike protein is replaced by the RBM from a pangolin strain, and in addition, a small but very special stretch of 4 amino acids is inserted, which creates a furin cleavage site that, as virologists have previously established, significantly expands the “repertoire” of the virus in terms of whose cells it can penetrate. Most likely, it was thanks to this new furin site that the new mutant managed to jump species from its original host to humans.
Indeed, virologists, including the leader of coronavirus research at the Wuhan Institute of Virology, Shi Zhengli, have done many similar things in the past — both replacing the RBM in one type of virus by an RBM from another, or adding a new furin site that can provide a species-specific coronavirus with an ability to start using the same receptor (e.g. ACE2) in other species. In fact, Shi Zhengli’s group was creating chimeric constructs as far back as 2007 and as recently as 2017, when they created a whole of 8 new chimeric coronaviruses with various RBMs. In 2019 such work was in full swing, as WIV was part of a $3.7 million NIH grant titled Understanding the Risk of Bat Coronavirus Emergence. Under its auspices, Shi Zhengli co-authored a 2019 paper that called for continued research into synthetic viruses and testing them in vitro and in vivo:
Currently, no clinical treatments or prevention strategies are available for any human coronavirus. Given the conserved RBDs of SARS-CoV and bat SARSr-CoVs, some anti-SARS-CoV strategies in development, such as anti-RBD antibodies or RBD-based vaccines, should be tested against bat SARSr-CoVs. Recent studies demonstrated that anti-SARS-CoV strategies worked against only WIV1 and not SHC014. In addition, little information is available on HKU3-related strains that have much wider geographical distribution and bear truncations in their RBD. Similarly, anti-S antibodies against MERS-CoV could not protect from infection with a pseudovirus bearing the bat MERSr-CoV S. Furthermore, little is known about the replication and pathogenesis of these bat viruses. Thus, future work should be focused on the biological properties of these viruses using virus isolation, reverse genetics and in vitro and in vivo infection assays. The resulting data would help the prevention and control of emerging SARS-like or MERS-like diseases in the future.
If the above quote might seem vague as to what exactly “using reverse genetics” might mean, the NIH grant itself spells it out:
Aim 3. In vitro and in vivo characterization of SARSr-CoV spillover risk, coupled with spatial and phylogenetic analyses to identify the regions and viruses of public health concern. We will use S protein sequence data, infectious clone technology, in vitro and in vivo infection experiments and analysis of receptor binding to test the hypothesis that % divergence thresholds in S protein sequences predict spillover potential.
“Infectious clone technology” stands for creating live synthetic viral clones. Considering the heights of user friendliness and automation that genetic engineering tools have attained, creating a synthetic CoV2 via the above methodology would be in reach of even a grad student.
But before delving into CoV2 origins, let’s first take a quick dive into its biology.
Biology
Ok, let’s start from the basics. What’s a furin site, an RBM, or a spike protein? Bear with me: once you wade through the jungle of terminology, conceptually, everything is pretty straightforward. For example, spike proteins are those red things sticking out of a virus particle — the very reason for which these viruses got “crowned”:
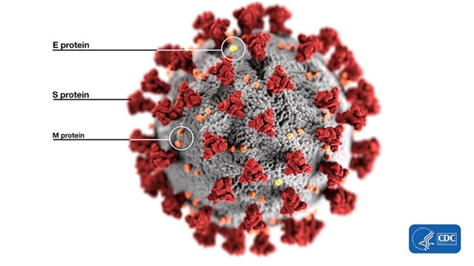
It is with the help of these proteins that the virion clings to the receptor of the victim cell (ACE2 in our case) to then penetrate inside. So it is a vitally important part of the virus, as without getting into a cell viruses cannot replicate. The spike protein also determines which animals the virus can or cannot infect, as ACE2 receptors (or other targets for other viruses) in different species can differ in structure. At the same time, out of the entire 30 kilobase genome (quite huge by viral standards), the gene of this protein makes up only 12–13%. So the spike protein is only about 1300 amino acids long. Below is how the spike (S) protein is structured in CoV2 and close relatives:
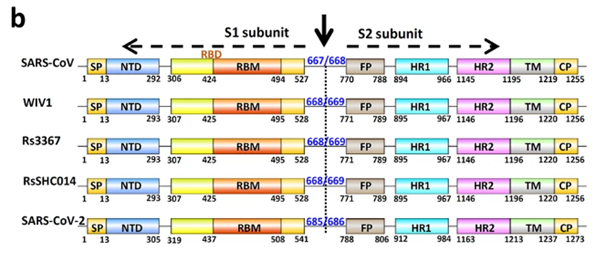
As can be seen from the figure above, the S protein consists of two subunits: S1 and S2. It is S1 that interacts with the ACE2 receptor, and the place where S1 does so is called Receptor Binding Domain (RBD), while the area of direct contact, the holy of holies, is called Receptor Binding Motif (RBM). Here is a beautiful illustration from an equally beautiful work:
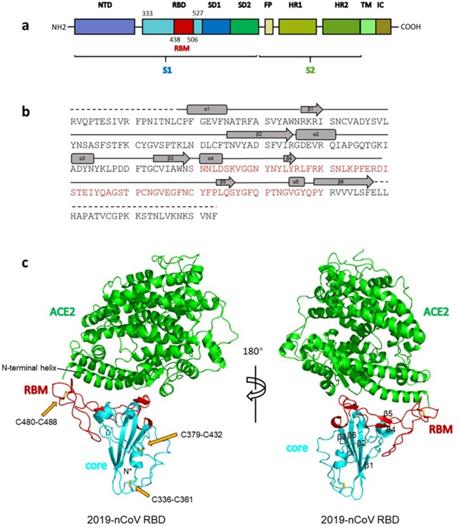
(a) Overall topology of 2019-nCoV spike monomer. NTD, N-terminal domain. RBD, receptor-binding domain. RBM, receptor-binding motif. SD1, subdomain 1. SD2, subdomain 2. FP, fusion peptide. HR1, heptad repeat 1. HR2, heptad repeat 2. TM, transmembrane region. IC, intracellular domain.
(b) Sequence and secondary structures of 2019-nCoV RBD. The RBM is colored red.
© Overall structure of 2019-nCoV RBD bound with ACE2. ACE2 is colored green. 2019-nCoV RBD core is colored cyan and RBM is colored red. Disulfide bonds in the 2019-nCoV RBD are shown as stick and indicated by yellow arrows. The N-terminal helix of ACE2 responsible for binding is labeled.
When the CoV2 genome was just sequenced and made publicly available on January 10, 2020, it was a riddle, as no closely related strains were known. But quite quickly, on January 23, Shi Zhengli released a paper indicating that CoV2 is 96% identical to RaTG13, a strain which her laboratory had previously isolated from Yunnan bats in 2013. However, outside of her lab, no one knew about that strain until January 2020.
It was immediately clear that RaTG13 is special. Take a look at the figure below:
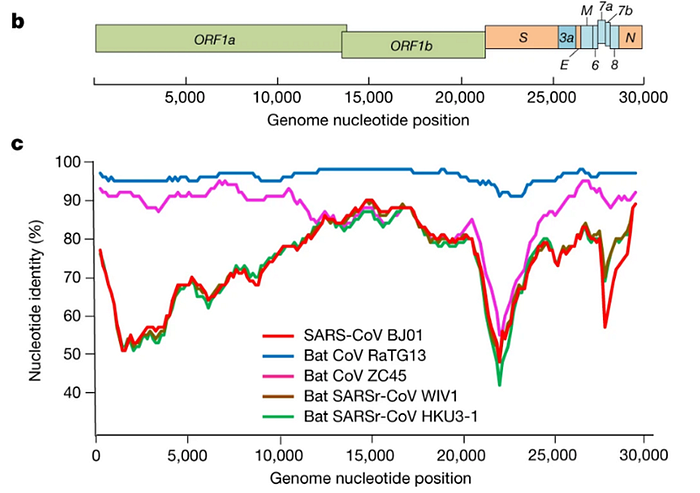
This is a genome similarity graph between CoV2 and other known strains. The higher the curve, the higher the percentage of matching nucleotides. As you can see, in the spike protein (S) gene region (between nucleotides 22k and 25k), only RaTG13 is more or less close to CoV2, while all other strains take a deep dive around this spot — both strains from other bats and the first SARS-CoV (red curve). This in itself is far from suspicious — who knows how many unknown SARS-like strains lurk in the bat caves of Yunnan? Ok, maybe it is not very clear how exactly the virus could get from there to Wuhan, but hey, with those wet markets you never know.
Pangolins
Next, pangolins appeared on the scene: in February, another group of Chinese scientists discovered a peculiar strain of pangolin coronavirus in their possession, which, while generally being only 90% similar to CoV2, in the RBM region was almost identical to it, with only a single amino acid difference (see the upper two sequences, dots indicate a match with the top sequence):
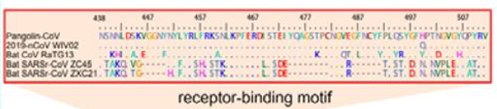
Surprisingly, in the first quarter of the S protein, the pangolin strain is highly dissimilar from CoV2, but after the RBM all three strains (CoV2, Pangolin, RaTG13) exhibit a shared high degree of similarity. Most strikingly, RaTG13’s RBM itself is quite different than that of CoV2, which can be seen from the steep dive of the green RaTG13 graph compared to the red CoV2 graph in the RBM region (pink strip) in the following graph:

This observation is confirmed by the phylogenetic analysis of the three areas highlighted in the graph above — in the RBM, the pangolin strain is closer to CoV2 than is RaTG13, but it is RaTG13 that is closer to CoV2 to the left and right of RBM. So there is obvious recombination, as the authors (and other papers) conclude.
How did the researchers obtain those pangolins? This is how:

They were confiscated from smugglers by Chinese customs and transferred to an animal rehab center in Guangdong, where they died while exhibiting severe coronavirus symptoms. This, of course, must have gotten the attention of local virologists, who took several samples:
Pangolins used in the study were confiscated by Customs and Department of Forestry of Guangdong Province in March-December 2019. They include four Chinese pangolins (Manis pentadactyla) and 25 Malayan pangolins (Manis javanica). These animals were sent to the wildlife rescue center, and were mostly inactive and sobbing, and eventually died in custody despite exhausting rescue efforts. Tissue samples were taken from the lung, lymph nodes, liver, spleen, muscle, kidney, and other tissues from pangolins that had just died for histopathological and virological examinations.
Those pangolins attracted the attention of other virologists too. For example, a team in Hong Kong also received samples of confiscated pangolins and in February 2020 they also released a paper that noted clear signs of recombination in the CoV2 spike protein:
We received frozen tissue (lungs, intestine, blood) samples that were collected from 18 Malayan pangolins (Manis javanica) during August 2017-January 2018. These pangolins were obtained during the anti-smuggling operations by Guangxi Customs. Strikingly, high-throughput sequencing of their RNA revealed the presence of coronaviruses in six (two lung, two intestine, one lung-intestine mix, one blood) of 43 samples. With the sequence read data, and by filling gaps with amplicon sequencing, we were able to obtain six full or nearly full genome sequences — denoted GX/P1E, GX/P2V, GX/P3B, GX/P4L, GX/P5E and GX/P5L — that fall into the 2019-CoV2 lineage (within the genus Betacoronavirus) in a phylogenetic analysis (Figure 1a).
…
More notable, however, was the observation of putative recombination signals between the pangolins coronaviruses, bat coronaviruses RaTG13, and human 2019-CoV2 (Figure 1c, d). In particular, 2019-CoV2 exhibits very high sequence similarity to the Guangdong pangolin coronaviruses in the receptor-binding domain (RBD; 97.4% amino acid similarity; indicated by red arrow in Figure 1c and Figure 2a), even though it is most closely related to bat coronavirus RaTG13 in the remainder of the viral genome. Bat CoV RaTG and the human 2019-CoV2 have only 89.2% amino acid similarity in RBD. Indeed, the Guangdong pangolin coronaviruses and 2019-CoV2 possess identical amino acids at the five critical residues of the RBD, whereas RaTG13 only shares one amino acid with 2019-CoV2 (residue 442, human SARS-CoV numbering).
By the way, the authors of this article also highlighted the high phylogenetic mosaicity of the CoV2 spike protein:
Interestingly, a phylogenetic analysis of synonymous sites alone in the RBD revealed that the phylogenetic position of the Guangdong pangolin is consistent with that in the remainder of the viral genome, rather than being the closest relative of 2019-CoV2 (Figure 2b). Hence, it is possible that the amino acid similarity between the RBD of the Guangdong pangolin coronaviruses and 2019-CoV2 is due to selectively-mediated convergent evolution rather than recombination, although it is difficult to choose between these scenarios on current data.
Translated from science-speak, what this means is that if we analyze the entire RBD of the three strains, ignoring the obvious differences (i.e. non-synonymous substitutions) among them, which are mainly found in the RBM (which, recall, is identical between CoV2 and Pangolin), and construct a phylogenetic tree for synonymous substitutions, CoV2 is still closer to RaTG13 than to the pangolin strain. Which is rather strange in light of the fact that the pangolin strain and CoV2 have identical RBMs (which are segments inside RBD).
The authors go on to put forth a conjecture that this may be the result of convergent evolution, in other words, that CoV2 and the pangolin strain came to possess identical RBMs each in their own way, rather than through recombination between common ancestors. Because it would have required a rather unique recombination event — as if someone cut out a precise RBM segment from a pangolin strain and used it to replace the RBM in RaTG13. Talk about Intelligent Design!
Royal Genealogy
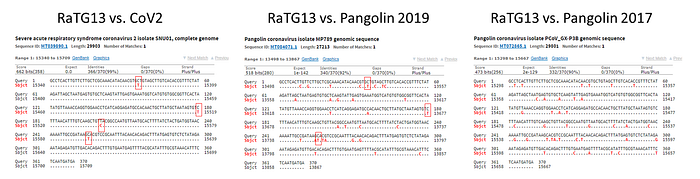
In order to better understand CoV2 origins, let’s take a look at spike protein sequences of our Unholy Trinity: CoV2, RaTG13 and MP789 (pangolin-2019). Let’s compare the pairwise differences between them (identical amino acids are marked with dots, red letters denote differences, and dashes indicate deleted/inserted amino acids):

The comparisons illustrate what previously quoted papers have noted: that in the first quarter of the sequence, the pangolin strain is far from CoV2 and RaTG1, and if it weren’t for the RBM region (red rectangle), RaTG13 would have been very close to CoV2. But, as I already said, the RBM in CoV2 is closest to that of the pangolin strain.
What about other pangolin strains? So far we’ve only analyzed the MP789 strain isolated from pangolins confiscated by customs in 2019. But there was another batch of pangolins confiscated in 2017, and they also had a similar coronavirus strain isolated. Let’s compare it to RaTG13 and MP789:

In the first quarter of the S protein, the 2017 pangolin strains are closer to RaTG13 (and CoV2) than their 2019 pangolin counterpart (MP789). At the same time, all three have a clear recent common ancestor in the areas marked by green rectangles, and in these areas RaTG13 and pangolin-2019 (MP789) are closer to each other than to pangolin-2017, since they have several common mutations (marked by red and blue ellipses), which are absent from pangolin-2017. But the RBM for all three is different, and different in approximately the same proportion, and in similar places.
Maybe after ancestors of RaTG13 and MP789 diverged, the MP789 ancestor had the first quarter of its protein replaced (which did not occur in RaTG13 or pangolin-2017), and the rest of the protein remained common for all three strains. Later the paths of the RaTG13 and MP789 gene pools crossed again and produced CoV2. It is also possible that the ancestor of RaTG13 arose as a result of recombination of ancestral pangolin strains.
It is also interesting to see a rather unique identical mutation (QTQTNS) in RaTG13 and pangolin-2019 right in front of the spot where CoV2 has a new furin cleavage site. That furin site, as I mentioned, arose via an insertion of 4 new amino acids (PRRA). If we look at the nucleotide sequence around this insertion, we can see that RaTG13 and CoV2 are closer to each other in that area than to pangolin-2019, since they possess several common mutations (highlighted in blue):

By the way, Orf1ab is also a phylogenetic mess in CoV2: 1a is closer to RaTG13, but 1b is closer to pangolin-2019:

Does this mean that the ancestor of CoV2 crossed with the common ancestor of pangolin-19 at least twice? First, when it (along with a common ancestor of RaTG13) inherited Orf1ab and the second half of the spike protein with the QTQTNS mutation, and second time when it acquired 1b and RBM, which differ from RaTG13. All of this is certainly possible in nature — after all, these viruses mutate and recombine constantly. Another question is where exactly bat and pangolin viruses are most likely to encounter one another for such orgies — in mountain caves, “wet markets”, shelters for confiscated animals, or even in laboratories. But let’s put those questions aside for now. First, let discuss what is arguably the most eye-catching aspect of the new virus — a 4-amino acid insertion that turned it into a natural-born killer.
A Killer Intro
It is impossible to ignore the introduction of a PRRA insert between S1 and S2: it sticks out like a splinter. This insert creates the furin cleavage site, which I mentioned at the very beginning. Let me explain what a furin site is. Remember the structure of our spike protein? Here is a detailed diagram:
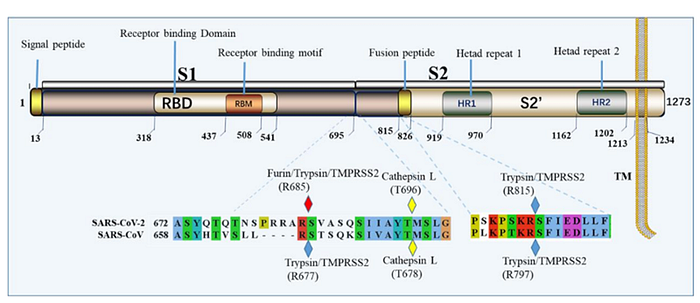
The protein consists of two parts, S1 and S2, of which S1 is responsible for primary contact with the receptor (recall Receptor Binding Domain / Motif), and S2 is responsible for fusion with the cell membrane and penetration into the cell. The fusion process is started by the fusion peptide marked in yellow, but in order for it to engage in its dirty deed, someone must cut the S protein at one of the sites marked by diamonds in the diagram above. The virus does not have its own such “cutters”, so it relies on various proteases of its victims. There are several types of such proteases, as can be deduced from the abundance of colors of those diamonds. But not all proteases are equal, and not all types of cells have proteases needed by the virus. Furin is one of the most effective, and it is found not only on the surface of cells, but also inside. Most clearly, the danger of the new furin site is demonstrated by the difference between CoV2 and its grandpa, SARS-CoV:
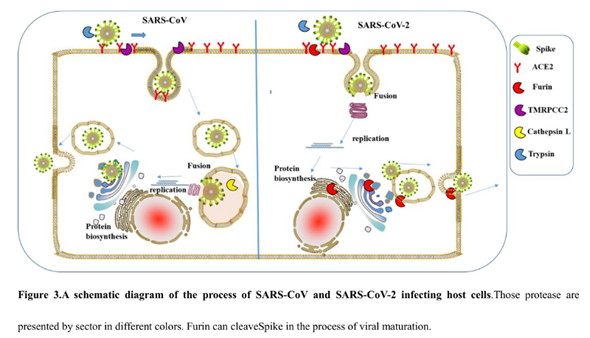
As can be seen from the diagram, in the case of CoV2, thanks to the furin site, it is not two, but three classes of proteases (three colored PacMans) that can cut its S protein outside the cell. But perhaps the most important difference is that furin is also present inside the cell, so it can cut the S protein immediately after virion assembly, thereby providing new virions with the ability to merge with new cells right off the bat (no pun intended).
The importance of the new furin site in CoV2’s virulence was recently demonstrated by a study in hamsters where the disappearance of the furin site (due to a mutation) greatly decreased mutant CoV2’s pathogenicity and replication ability:
Infection of hamsters shows that one of the variants (Del-mut-1) which carries deletion of 10 amino acids (30 bp) does not cause the body weight loss or more severe pathological changes in the lungs that is associated with wild type virus infection.
The good news is that there already exist various furin and other protease inhibitors, and some of them (like camostat and its analogs) are already being clinically tested against CoV2.
By the way, it is possible that the new furin site could also be largely responsible for the pronounced age-dependent morbidity and mortality of CoV2:
Patients with hypertension, diabetes, coronary heart disease, cerebrovascular illness, chronic obstructive pulmonary disease, and kidney dysfunction have worse clinical outcomes when infected with SARS-CoV-2, for unknown reasons. The purpose of this review is to summarize the evidence for the existence of elevated plasmin(ogen) in COVID-19 patients with these comorbid conditions. Plasmin, and other proteases, may cleave a newly inserted furin site in the S protein of SARS-CoV-2, extracellularly, which increases its infectivity and virulence.
Furin cuts proteins in strictly defined places, namely after an RxxR sequence (that is, Arg-X-X-Arg, where X can be any amino acid). Moreover, if arginine is also in the second or third place (that is, RRxR or RxRR), then the cleavage efficiency is significantly increased.
Therefore, the appearance of a new furin cleavage site was noticed immediately, as none of the closest or even distant relatives of Cov2 have such a site — those coronaviruses that do, share only 40% of their genome with Cov2:
It was found that all Spike with a SARS-CoV-2 Spike sequence homology greater than 40% did not have a furin cleavage site (Figure 1, Table 1), including Bat-CoV RaTG13 and SARS-CoV (with sequence identity as 97.4% and 78.6%, respectively). The furin cleavage site “RRAR” in SARS-CoV-2 is unique in its family, rendering by its unique insert of “PRRA”. The furin cleavage site of SARS-CoV-2 is unlikely to have evolved from MERS, HCoV-HKU1, and so on. From the currently available sequences in databases, it is difficult for us to find the source. Perhaps there are still many evolutionary intermediate sequences waiting to be discovered.
Here is a great illustration from the source article of the quote above. Coronaviruses with a furin site are marked in pink, 3 different strains of Cov2 are shown at 10 o’clock:
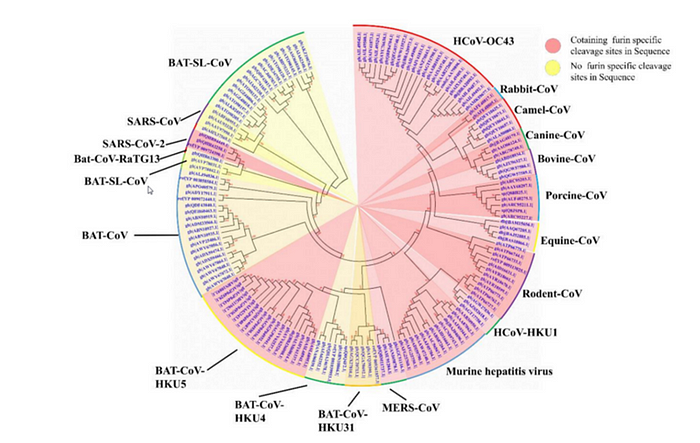
The closest relative with a furin site is the HKU5 strain, isolated by the Shi Zhengli team in 2014 in Guangzhou from bats of the genus Pipistrellus (added to GenBank in 2018). But it is a very distant relative — their spike proteins share only 36%.
So the virologists are puzzled. Where did this 12 nucleotide insert come from? Could it be lab-made? Well, virologists have studied furin sites in coronaviruses for decades, and have introduced many artificial ones in a lab. For example, an American team had inserted RRSRR into the spike protein of the first SARS-CoV back in 2006:
To investigate whether proteolytic cleavage at the basic amino acid residues, were it to occur, might facilitate cell–cell fusion activity, we mutated the wild-type SARS-CoV glycoprotein to construct a prototypic furin recognition site (RRSRR) at either position.

And the Japanese have inserted a similar site (RRKR) into the SARS-CoV protein in 2008, though a bit downstream than in CoV2:
In the same year 2008, their Dutch colleagues also studied these protease sites of SARS-CoV and compared them to the murine coronavirus MHV, which also has such a site (SRRAHR | SV), one that is quite similar to the site of CoV2 (SPRRAR | SV):
In 2009, another American group also worked on “improving” SARS-CoV and, continuing the American tradition of not penny-pinching on arginines, they inserted as many as 4 of them (RRSRR):
To examine the potential use of the SARS-CoV S1–S2 and S2′ positions as sites for proteolytic cleavage, we first introduced furin cleavage recognition sites at these locations by making the following mutations 664-SLLRSTSQSI — SLLRRSRRSI-671 (S1–S2) and 792-LKPTKRSF — LKRTKRSF-799 (S2′).
Beijing 2019
But the most recent work of this kind that I came across was an October 2019 paper from several Beijing labs, where the new furin site RRKR was inserted into not just some pseudovirus, but into an actual live chicken coronavirus, infectious bronchitis virus (IBV):
An interesting side note is that, as the authors point out, the addition of a furin site allows the mutant virus to infect nerve cells. Perhaps the CoV2 furin site is the reason why some patients with CoV2 exhibit neurological symptoms, including loss of smell:
Mutation of the S2' site of QX genotype (QX-type) spike protein (S) in a recombinant virus background results in higher pathogenicity, pronounced neural symptoms and neurotropism when compared with conditions in wild-type IBV (WT-IBV) infected chickens. In this study, we present evidence suggesting that recombinant IBV with a mutant S2' site (furin-S2' site) leads to higher mortality. Infection with mutant IBV induces severe encephalitis and breaks the blood–brain barrier.
…
In summary, our results demonstrate that the furin cleavage site upstream of the FP in S protein is an important site for CoV, modulating entry, cell–virus fusion, adaptation to its host cell, cell tropism and pathogenicity, but not antigenicity.
To be clear, many coronaviruses have naturally occurring furin sites, and they are very diverse. Obviously, they can appear as a result of random mutations. This is what happened in the case of MERS, as was pointed out in 2015 by an international team of authors, including Shi Zhengli and Ralph Baric, two stars of synthetic coronavirusology. We will come back to them many times, but for now, a few words about that article. In it the authors have shown that just two mutations allowed MERS to jump from bats to humans, and one of these mutations created a furin site. Though it was not an insertion of new amino acids, but a mutation of an existing one (marked in red on the left below):
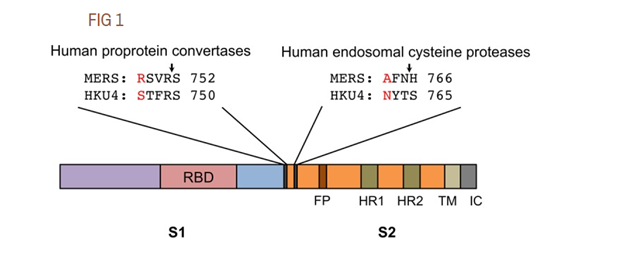
The authors did not just show this, but actually introduced these mutations back into the original bat strain: they created the same furin site and showed that it enables the bat strain to infect human cells:
To evaluate the potential genetic changes required for HKU4 to infect human cells, we reengineered HKU4 spike, aiming to build its capacity to mediate viral entry into human cells. To this end, we introduced two single mutations, S746R and N762A, into HKU4 spike. The S746R mutation was expected to restore the hPPC motif in HKU4 spike, whereas the N762A mutation likely disrupted the potential N-linked glycosylation site in the hECP motif in HKU4 spike.
…
We examined the capability of the mutant HKU4 spike to mediate viral entry into three types of human cells (Fig. 3A for HEK293T cells; data not shown for Huh-7 and MRC-5 cells), using a pseudovirus entry assay as previously described (14). In the absence of exogenous protease trypsin, HKU4 pseudoviruses bearing either the reengineered hPPC motif or the reengineered hECP motif were able to enter human cells, whereas HKU4 pseudoviruses bearing both of the reengineered human protease motifs entered human cells as efficiently as when activated by exogenous trypsin (Fig. 3A). In contrast, wild-type HKU4 pseudoviruses failed to enter human cells. Therefore, the reengineered hPPC and hECP motifs enabled HKU4 spike to be activated by human endogenous proteases and thereby allowed HKU4 pseudoviruses to bypass the need for exogenous proteases to enter human cells. These results reveal that HKU4 spike needs only two single mutations at the S1/S2 boundary to gain the full capacity to mediate viral entry into human cells.
By the way, how they did it might frighten those who aren’t familiar with modern biotechnology — because the authors inserted this coronavirus spike-like protein into inactivated HIV:
Briefly, MERS-CoV-spike-pseudotyped retroviruses expressing a luciferase reporter gene were prepared by cotransfecting HEK293T cells with a plasmid carrying Env-defective, luciferase-expressing HIV-1 genome (pNL4–3.luc.R-E-) and a plasmid encoding MERS-CoV spike protein.
Perhaps this is what prompted Indian researchers to look for sequences similar to HIV in the CoV2 genome (but their preprint was quickly criticized for bad methodology and erroneous conclusions). In fact, experts use such pseudoviruses regularly, and in general, one should not be scared of retroviruses as a class — their subspecies lentiviruses have been used for gene therapy for many years.
Where Did RaTG13 Come From?
RaTG13 is a very unusual strain. Odd to see that Shi Zhengli’s group was silent about it for all these years. After all, it is very different from its SARS-like siblings, especially in the spike protein, which is precisely what determines which types of cells (and in which animals) this virus can infect. Here is a genome similarity graph of CoV2 compared to other bat coronaviruses (panel B):
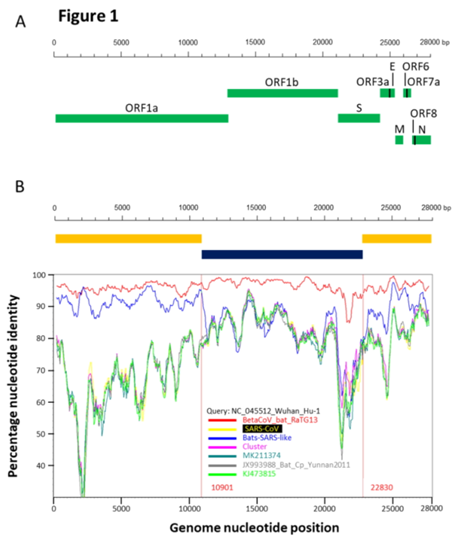
The red curve represents RaTG13 while the blue curve is for the strains closest to RaTG13 (ZXC21 and ZC45). These strains were isolated from Chinese horseshoe bats (Rhinolophus sinicus) in Zhoushan in 2015 (ZXC21) and 2017 (ZC45). As can be seen from the above graph, even they differ in their S proteins from RaTG13. A direct sequence comparison illustrates this difference best:
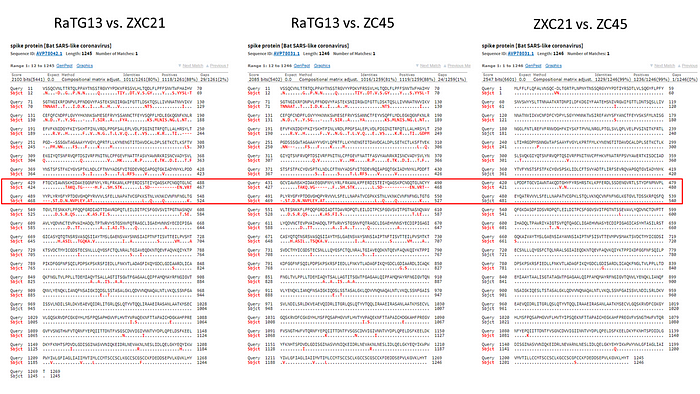
As we can see, the spike proteins of ZXC21 and ZC45 are not only 23–24 amino acid residues shorter than the RaTG13 protein, but they are shorter in the most important place — in the RBM (note the deletions in the red box marked with red dashes).
So where did RaTG13 come from? As I already mentioned, in 2020 Shi Zhengli reported that she isolated it in 2013 from Yunnan horseshoe bats (from Rhinolophus affinis, not the usual suspects R. sinicus). But until January 2020, this strain’s existence was not known, and here is how Shi Zhengli’s group described their discovery about RaTG13’s similarity to CoV2:
We then found that a short region of RNA-dependent RNA polymerase (RdRp) from a bat coronavirus (BatCoV RaTG13) — which was previously detected in Rhinolophus affinis from Yunnan province — showed high sequence identity to 2019-CoV2. We carried out full-length sequencing on this RNA sample (GISAID accession number EPI_ISL_402131). Simplot analysis showed that 2019-CoV2 was highly similar throughout the genome to RaTG13 (Fig. 1c), with an overall genome sequence identity of 96.2%.
Not much detail: previously detected, and that is that. Moreover, the quote seems to imply that until 2020, they only sequenced a part of its genome, the RdRp gene (which is part of Orf1b that precedes the spike protein gene). Ok, but where exactly in Yunnan was it obtained? The paper doesn’t mention it, and neither does GenBank. However, the GISAID entry seems to have a bit more info: collected in Pu’er City from a male bat’s fecal swab:

This rang a bell, as in my wanderings around Pubmed, I had already encountered an expedition to Pu’er in the summer of 2013:
Bats were captured from various locations in five counties of four prefectures of Yunnan Province, China, from May to July 2013.
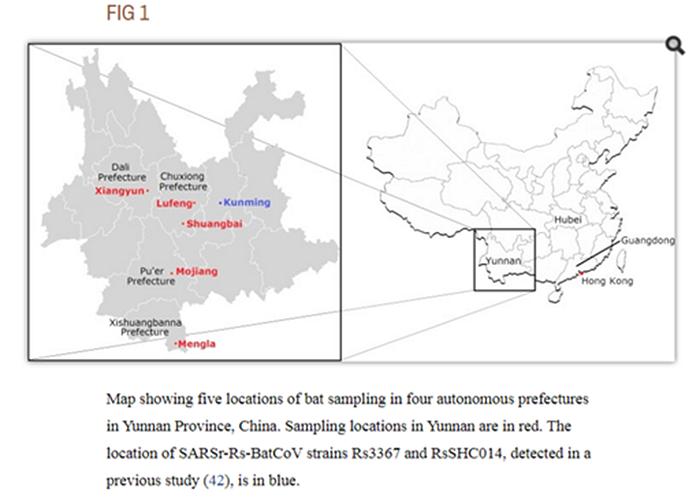
Researchers did not report anything particularly interesting for us from that expedition, but maybe it was then that Shi Zhengli or someone from her group obtained the RaTG13 sample? Which they sequenced only partially, and for some reason decided not to publish, although it was very different from everything known before.
By the way, Shi Zhengli could well have personally participated in that expedition, as she expressed great fondness when describing them — for example, in her TED-like talk in 2018, where she showed personal photos from such expeditions:
Moreover, it was a series of exactly such expeditions that brought Shi Zhengli worldwide fame and a “Batwoman” moniker: in a 2013 Nature paper, her group triumphantly announced that in Yunnan caves they had discovered carrier bats of the RsSHC014 and Rs3367 strains that coincided with the first SARS-CoV by 85% and 96%, respectively.
It is quite a coincidence that around the same time in Yunnan, Shi Zhengli’s group also discovered RaTG13, the closest strain to CoV2, and the two also share 96% of their genomes.
UPD: Is RaTG13 the same as RaBtCoV/4991?
[UPDATED] After I had published this post, I was pointed to this preprint that alleges that RaTG13 is, in fact, RaBtCoV/4991 (KP876546), which Shi Zhengli had previously reported discovering in an abandoned mineshaft in Yunnan in 2013. There indeed are several reasons to think so. First and foremost, the only published sequence for RaBtCoV/4991 is 100% identical to that of RaTG13 at the nucleotide level, albeit being just a 370-bp stretch of the RdRp gene:
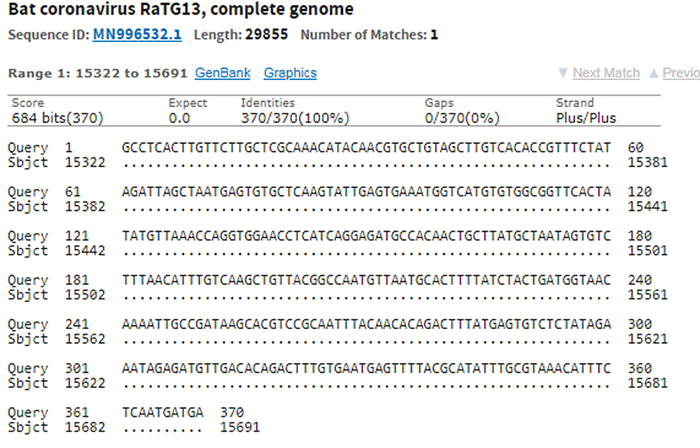
Second, the collection details of the two strains are nearly identical: both were collected in July 2013 from a fecal swab of R. affinis bats:

RaBtCoV/4991 was collected in a mineshaft located in the Mojiang county, which is under the jurisdiction of Pu’er City:
Mojiang Hani Autonomous County is an autonomous county under the jurisdiction of Pu’er City, in the south of Yunnan Province, China.
Wikipedia
And Pu’er City is listed as the collection location of RaTG13 at the GISAID database, which could well be an approximation for the Mojiang mineshaft.
It is odd that in her 2020 paper on RaTG13 Shi Zhengli fails to mention RaBtCoV/4991 or cite her 2016 paper about its discovery, for which she is listed as the one who “designed and coordinated the study”. It is not like RaBtCoV/4991 was forgotten by her group, as it is mentioned in their 2019 paper, where it is included in a phylogenetic tree of other coronaviruses:

I doubt that RaBtCoV/4991’s place in that tree was determined based solely on a 370-bp fragment, so I would think that by early 2019, Shi Zhengli’s group would have sequenced its full genome.
Intriguingly, both pangolin-2017 and pangolin-2019 genomes are also very close in this stretch of the RdRp gene, and CoV2 and pangolin-2019 share a few common mutations not found in RaTG13:
But let’s put this topic aside for now and get back to the story of Shi Zhengli’s famous 2013 Nature paper.
“Wuhan-1”
In that paper, Shi Zhengli’s group also reported that by culturing the isolated samples in monkey Vero cells, they managed to isolate a live virus that was almost identical to the Rs3367 strain. The authors named their creation WIV1 (where WIV stands for Wuhan Institute of Virology):
Most importantly, we report the first recorded isolation of a live SL-CoV (bat SL-CoV-WIV1) from bat faecal samples in Vero E6 cells, which has typical coronavirus morphology, 99.9% sequence identity to Rs3367 and uses ACE2 from humans, civets and Chinese horseshoe bats for cell entry. Preliminary in vitro testing indicates that WIV1 also has a broad species tropism.
Let’s compare RaTG13 with Rs3367 and RsSHC014:

As we can see, the spike proteins of these strains are not only 13 amino acids shorter than that of RaTG13, but they also differ in the first quarter of the protein quite substantially. By the way, it is curious that the spike proteins of Rs3367 (aka WIV1) and RsSCH014 are almost identical, and differ only in the RBD region (right sequence below). Almost like CoV2 and RaTG13 (not counting the furin insert):

Could researchers, having received coronavirus samples from pangolins that were intercepted by customs in March 2019, then want to check whether the RBM in pangolin strains can bind to the human ACE2 receptor? And could such researchers also decide to throw an extra furin site in the mix?
Theoretically, of course, they could. From a technical standpoint, it is almost routine for virologists to conduct such experiments. A reasonable question might be: why use RaTG13 as a backbone, and not, say, the tried and true WIV1? Well, it doesn’t have to be either-or: maybe a chimera with WIV1 was also tested. But in parallel, they might have decided to simulate recombination of the pangolin virus with the bat strain closest to it — after all, RaTG13 is much closer to the pangolin strains than WIV1: its spike protein is closer to them both phylogenetically and structurally — it even matches them in length, while the proteins of WIV1/Rs3367 and RsSHC014 are 13 amino acids shorter. Also, the QTQTNS mutation common to RaTG13 and pangolin-2019 (MP789) just before the protease site could not have gone unnoticed by coronavirus experts.
Other Yunnan Strains
In 2011, other researchers had also found samples of coronaviruses from the Yunnan Rhinolophus affinis. The strain LYRa11 seemed to me the most interesting:

But it is also quite distant from RaTG13, and much closer to Rs3367 (that’s the strain that shares 96% with the first SARS-CoV):

But RaTG13, isolated from the same Rhinolophus affinis bats as LYRa11, looks the least like it (left sequence comparison).
Finally, another Yunnan strain (ingenuously named Yunnan2011), isolated in 2011 from another subspecies of horseshoe bats, Rhinolophus pusillus, is even less similar to RaTG13 than LYRa11:

Between themselves, Yunnan2011 and LYRa11 (the right sequence above) are not particularly similar, apart from the highly conserved S2 region. By the way, what’s up with the differing naming conventions for these strains? Sometimes they fully spell out the year, sometimes partially, yet other times not at all (Rs3367). The carrier species sometimes leads (RaTG13), sometimes follows (LYRa11). And what do TG, LY or SHC stand for? Initials of the person sequencing the genome?
Anyways, let’s move on from viral archeology to viral engineering, namely transplanting key areas of the spike protein between species and other gain-of-function (GOF) experiments.
1999: First Chimeric Coronavirus
If you think that all of the gain-of-function coronavirus research into what exactly allows coronaviruses to jump from one species to another began in response to the first SARS outbreak in 2002, you’d be mistaken. Virologists experimented with chimeric coronaviruses long before that. Here, for example, is a 1999 paper from the Dutch group of Peter Rottier from Utrecht University with a revealing title Retargeting of Coronavirus by Substitution of the Spike Glycoprotein Ectodomain: Crossing the Host Cell Species Barrier:
Using targeted RNA recombination, we constructed a mutant of the coronavirus mouse hepatitis virus (MHV) in which the ectodomain of the spike glycoprotein (S) was replaced with the highly divergent ectodomain of the S protein of feline infectious peritonitis virus. The resulting chimeric virus, designated fMHV, acquired the ability to infect feline cells and simultaneously lost the ability to infect murine cells in tissue culture.
By the way, Shi Zhengli seems to have worked under the supervision of Peter Rottier in Utrecht for a time. At least in 2005, she co-authored a joint paper where Utrecht was listed as her affiliation (but her current address was listed at Shanghai Institute). [CORRECTION: The italicized text is incorrect, it was not Shi Zhengli but a different person]. That article itself is quite curious — in it the authors investigated what exactly allows viruses to expand their species tropism:
Only a relatively few mutations in its spike protein allow the murine coronavirus to switch from a murine-restricted tropism to an extended host range by being passaged in vitro. One such virus that we studied had acquired two putative heparan sulfate-binding sites while preserving another site in the furin-cleavage motif. The adaptation of the virus through the use of heparan sulfate as an attachment/entry receptor was demonstrated by increased heparin binding as well as by inhibition of infection through treatment of cells and the virus with heparinase and heparin, respectively.
It is interesting that the furin site in that virus (SRRAHR | SV) is similar to the site in CoV2 (SPRRAR | SV), although in CoV2 it is cut more efficiently due to dual arginines (this is what makes it a polybasic site, i.e. it has multiple basic amino acids in a row in the RxxR sequence):
But what is especially curious is that the mutations that allowed the virus to “expand its horizons” occurred not in animals, but in vitro. Moreover, it seems, they happened pretty quickly:
MHV/pi23, a virus obtained after 23 of the 600 passages that resulted in MHV/BHK, also contains a putative HS-binding site in the S1 domain at the same position as in MHV/BHK, albeit as a smaller insertion, while it lacks the putative HS-binding site immediately upstream of the fusion peptide. MHV/pi23 does infect nonmurine cells to some extent but much less efficiently than MHV/BHK. In addition to the multiple HS-binding sites, however, mutations found in other parts of the S protein, such as the HR1 domain and the putative fusion peptide (Fig. 1), might also contribute to the efficient entry into nonmurine cells. We are currently in the process of determining the S protein mutations that are required for the extended host range phenotype.
Skipping ahead, I’ll just mention that there were other groups that used in vitro mutagenesis to increase the virulence of coronaviruses, for example, MERS:
To better understand the species adaptability of MERS-CoV, we identified a suboptimal species-derived variant of DPP4 to study viral adaption. Passaging virus on cells expressing this DPP4 variant led to accumulation of mutations in the viral spike which increased replication.
Moreover, their mutations arose after just several passages (rounds of cell culture reproduction):
(G) Location of mutations within MERS-CoV spike.
But those experiments occurred much later. In the meantime, let’s go back to 2002 — BEFORE the outbreak of the first SARS-CoV.
Ralph “Trailblazer” Baric
Ralph Baric is a legend in coronavirology. He is a trailblazer of synthetic genomic manipulation techniques. Back in 2002, he published a breakthrough work, which marked a milestone in both the study of various mechanisms of natural viruses and in gain-of-function research. In their paper, the Baric group described creating a synthetic clone of a natural murine coronavirus:
A novel method was developed to assemble a full-length infectious cDNA of the group II coronavirus mouse hepatitis virus strain A59 (MHV-A59). Seven contiguous cDNA clones that spanned the 31.5-kb MHV genome were isolated. The ends of the cDNAs were engineered with unique junctions and assembled with only the adjacent cDNA subclones, resulting in an intact MHV-A59 cDNA construct of ∼31.5 kb in length. The interconnecting restriction site junctions that are located at the ends of each cDNA are systematically removed during the assembly of the complete full-length cDNA product, allowing reassembly without the introduction of nucleotide changes… The method has the potential to be used to construct viral, microbial, or eukaryotic genomes approaching several million base pairs in length and used to insert restriction sites at any given nucleotide in a microbial genome.
In essense, the authors have “translated” the RNA virus into the language of DNA (using reverse transcriptase), which enabled them to manipulate its genome with the help of existing genetic engineering tools. Having created 7 such cDNA provirus segments, the authors then stitched them together “seamlessly” (i.e. without introducing any new, even silent mutations, including new restrictase sites), after which they transcribed their construct back into RNA, which was then translated into virus particles in other cells.
SARS-2003
Just a few weeks after the publication of the above work, the first SARS-CoV epidemic broke out. The Baric group sprang into action. By summer of 2003, they have submitted a paper on synthetically recreating SARS-CoV:
Using a panel of contiguous cDNAs that span the entire genome, we have assembled a full-length cDNA of the SARS-CoV Urbani strain, and have rescued molecularly cloned SARS viruses (infectious clone SARS-CoV) that contained the expected marker mutations inserted into the component clones. Recombinant viruses replicated as efficiently as WT virus and both were inhibited by treatment with the cysteine proteinase inhibitor… Availability of a SARS-CoV full-length cDNA provides a template for manipulation of the viral genome, allowing for the rapid and rational development and testing of candidate vaccines and therapeutics against this important human pathogen.
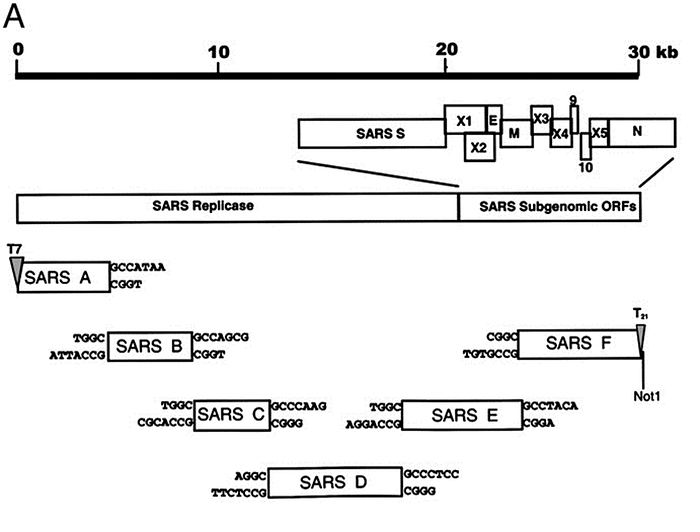
The speed of the Baric group illustrates how quickly a qualified team of virologists can create a synthetic clone from a natural virus, and therefore make genetic modifications to it. Moreover, that was back in 2003. Today, a qualified laboratory can repeat those steps in a matter of weeks.
In fact, two just did: the Swiss have created a synthetic clone of CoV2 in under a month, while it took the Galveston BSL4 lab less than 2 months to do so.
SARS-2006
Baric was the first, but far from the last. Genetic engineering developed by leaps and bounds, creating newer and better tools. Other groups explored alternative synthetic virology techniques. For example, in 2006, Spanish researchers followed in Baric’s footsteps, also creating a synthetic SARS clone, but using an alternative approach (bacterial artificial chromosome):
The engineering of a full-length infectious cDNA clone and a functional replicon of the severe acute respiratory syndrome coronavirus (SARS-CoV) Urbani strain as bacterial artificial chromosomes (BACs) is described in this study. In this system, the viral RNA was expressed in the cell nucleus under the control of the cytomegalovirus promoter and further amplified in the cytoplasm by the viral replicase. Both the infectious clone and the replicon were fully stable in Escherichia coli.
…
The assembled SARS-CoV infectious cDNA clone was fully stable during its propagation in E. coli DH10B cells for more than 200 generations, considerably facilitating the genetic manipulation of the viral genome (data not shown). The detailed cloning strategy, plasmid maps, and sequences are available upon request.
(A) Genetic structure of the SARS-CoV Urbani strain genome. Relevant restriction sites used for the assembly of the full-length cDNA clone are indicated. Numbers in parentheses indicate the genomic positions of the first nucleotide of the restriction endonuclease recognition sequence. Letters and numbers indicate the viral genes. L, leader sequence; UTR, untranslated region; An, poly(A) tail. (B) Construction of pBAC-SARS-CoV 5′-3′. After the selection of appropriate restriction sites, the intermediate plasmid pBAC-SARS-CoV 5′-3′ was constructed as the backbone for assembling the infectious cDNA clone. This plasmid includes the first 681 nt of the genome under the control of the CMV promoter, a multiple-cloning site containing the restriction sites selected for the final assembly of the infectious clone, and the last 975 nt of the genome, followed by a synthetic poly(A) tail (pA), the hepatitis delta virus ribozyme (Rz), and the bovine growth hormone termination and polyadenylation sequences (BGH). All these elements were precisely joined by overlapping PCR. The CMV promoter transcription start and the ribozyme cleavage site are shown. © Schematic diagram showing the five-step cloning strategy used for the assembly of the SARS-CoV full-length cDNA clone. The five overlapping cDNA fragments, named SARS 1 to SARS 5, were sequentially cloned into the plasmid pBAC-SARS-CoV 5′-3′ to generate the plasmid pBAC-SARS-CoVFL. Relevant restriction sites are indicated. The labels are as described for panel A.
True, they didn’t do it as elegantly as Baric, as their final assembly of the synthetic virus included their added restriction enzyme sites, while Baric learned to combine fragments “seamlessly”. But this is a minor point, the Spanish approach is just as robust — in 2013, with its help, the same authors had created a synthetic clone of MERS, and in 2015 their technique was included in a coronavirus textbook (chapter 13).
Wuhan 2007
Let’s get back to 2007. That is when the Shi Zhengli group joined the synthetic virology race with a study of the spike protein of human and bat coronaviruses, trying to determine what exactly is responsible for the ability to skip from one species to another:
A series of S chimeras was constructed by inserting different sequences of the SARS-CoV S into the SL-CoV S backbone.
That is, the authors inserted different segments from the human SARS-CoV spike protein into the spike protein of the bat virus. Here is their conclusion:
From these results, it was deduced that the region from aa 310 to 518 of BJ01-S was necessary and sufficient to convert Rp3-S into a huACE2-binding molecule.
At the same time, they tried to replace shorter fragments, including just the RBM:
For introduction of the RBM of SARS-CoV S into the SL-CoV S, the coding region from aa 424 to 494 of BJ01-S was used to replace the corresponding regions of Rp3-S, resulting in a chimeric S (CS) gene designated CS424–494.
Given that the above was written in 2007, I think today it will not be difficult for even a novice virologist to replace the RBM of one virus by an RBM from another.
Chimera-2015
In light of the above experiments, it is not very clear what caused the uproar that followed probably the most famous gain-of-function virology paper. I am referring to the joint 2015 work of Shi Zhengli and Ralph Baric, in which they created a synthetic chimeric virus:
Using the SARS-CoV reverse genetics system, we generated and characterized a chimeric virus expressing the spike of bat coronavirus SHC014 in a mouse-adapted SARS-CoV backbone. The results indicate that group 2b viruses encoding the SHC014 spike in a wild-type backbone can efficiently use multiple orthologs of the SARS receptor human angiotensin converting enzyme II (ACE2), replicate efficiently in primary human airway cells and achieve in vitro titers equivalent to epidemic strains of SARS-CoV. Additionally, in vivo experiments demonstrate replication of the chimeric virus in mouse lung with notable pathogenesis. Evaluation of available SARS-based immune-therapeutic and prophylactic modalities revealed poor efficacy; both monoclonal antibody and vaccine approaches failed to neutralize and protect from infection with CoVs using the novel spike protein. On the basis of these findings, we synthetically re-derived an infectious full-length SHC014 recombinant virus and demonstrate robust viral replication both in vitro and in vivo.
To me, the authors followed a familiar path: they took the spike-like protein from RsSHC014, which Shi Zhengli isolated from Yunnan bats in 2011, and inserted it into a murine-adapted variant of SARS-CoV for subsequent in vivo experiments. They also tested it in human cells, and almost as an aside created a recombinant clone of the same RsSHC014 strain:
The researchers also uncovered that it was not only the binding of spike protein to the receptor that determined the virus’s potential for transition from one animal species to another, because the SHC014-MA15 chimera was more virulent than SHC014 itself, even in human cells:
Notably, differential tropism in the lung as compared to that with SARS-MA15 and attenuation of full-length SHC014-CoV in [human epithelial airway cell] cultures relative to SARS-CoV Urbani suggest that factors beyond ACE2 binding — including spike processivity, receptor bio-availability or antagonism of the host immune responses — may contribute to emergence.
I especially want to highlight the spike processivity in the quote, because this is not the first time that virologists have mentioned that the ability of a spike protein to be cleaved by proteases (including furin) can have an impact on virulence.
That’s all I have to say about that paper. As a curiosity here is a common photo of its key authors, which was taken in Wuhan, in October 2018. Fittingly, Ralph Bariс and Shi Zhengli are front and center. I call this photo “The Wuhan Clan”. (Sorry, couldn’t resist).

Murine SARS-2007
One quick aside regarding the “murine virus MA15” from the above paper. That was not some kind of natural murine coronavirus, as one might think. It was a laboratory-modified human SARS-CoV, which back in 2007 the Baric group — possibly in competition with the Shi Zhengli group (remember their article from 2007) — turned into a real beast. To do this, they first iteratively “improved” it in mice, and when after several iterations it became maximally “effective”, they reproduced the observed mutations in a synthetic clone, and once again checked that it really does have increased virulence and lethality:
We adapted the SARS-CoV (Urbani strain) by serial passage in the respiratory tract of young BALB/c mice. Fifteen passages resulted in a virus (MA15) that is lethal for mice following intranasal inoculation. Lethality is preceded by rapid and high titer viral replication in lungs, viremia, and dissemination of virus to extrapulmonary sites accompanied by lymphopenia, neutrophilia, and pathological changes in the lungs. Abundant viral antigen is extensively distributed in bronchial epithelial cells and alveolar pneumocytes, and necrotic cellular debris is present in airways and alveoli, with only mild and focal pneumonitis. These observations suggest that mice infected with MA15 die from an overwhelming viral infection with extensive, virally mediated destruction of pneumocytes and ciliated epithelial cells. The MA15 virus has six coding mutations associated with adaptation and increased virulence; when introduced into a recombinant SARS-CoV, these mutations result in a highly virulent and lethal virus (rMA15), duplicating the phenotype of the biologically derived MA15 virus. Intranasal inoculation with MA15 reproduces many aspects of disease seen in severe human cases of SARS.
Baric-2008
Here is another example of the potential scientific rivalry between the Baric and Shi Zhengli groups. In 2008, the Baric group took the Bat-SCoV strain and replaced its RBD with an RBD from human SARS. That is, they essentially reproduced the work of Shi Zhengli’s group from 2007, except they didn’t limit themselves to pseudo-viruses, but created a real chimeric virus:
Here, we report the design, synthesis, and recovery of the largest synthetic replicating life form, a 29.7-kb bat severe acute respiratory syndrome (SARS)-like coronavirus (Bat-SCoV), a likely progenitor to the SARS-CoV epidemic.
…
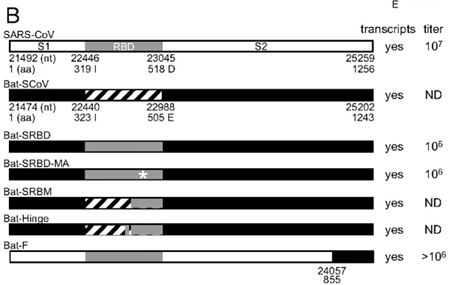
To test whether the RBDs of Bat-SCoV and SARS-CoV were interchangeable, we replaced the Bat-SCoV RBD (amino acid 323–505) with the SARS-CoV RBD (amino acid 319–518) (27, 28) (GenBank accession no. FJ211860), simulating a theoretical recombination event that might occur during mixed infection in vivo (Fig. 1B).
Baric-2016
The Baric group does seem to have its share of similar papers. For example, in 2016, they essentially repeated their collaboration with Shi Zhengli from 2015 to create a chimeric virus, only this time they inserted a spike protein segment into their mouse-adapted SARS not from RsSCH014, but from another strain Shi Zhengli found in Yunnan — its close relative Rs3367. Or, to be exact, from WIV1 — the laboratory clone of Rs3367 isolated at the Wuhan Institute of Virology in 2013:
Using the SARS-CoV infectious clone as a template (7), we designed and synthesized a full-length infectious clone of WIV1-CoV consisting of six plasmids that could be enzymatically cut, ligated together, and electroporated into cells to rescue replication competent progeny virions (Fig. S1A). In addition to the full-length clone, we also produced WIV1-CoV chimeric virus that replaced the SARS spike with the WIV1 spike within the mouse-adapted backbone (WIV1-MA15, Fig. S1B). … To confirm growth kinetics and replication, Vero cells were infected with SARS-CoV Urbani, WIV1-MA15, and WIV1-CoV.
To me, the 2016 paper looks a lot like the 2015 one. Moreover, its rationale is not very clear to me: after all, WIV1/Rs3367 already shared 96% of their genome with SARS-CoV. So I am not sure why one would want to insert a spike protein from its closest relative back into SARS-CoV. Maybe just because they could. In this light, the title of the article acquires a certain duality: SARS-like WIV1-CoV poised for human emergence.
Oh, and I am not sure how in 2015 Baric was granted a patent for the creation of “chimeric coronavirus spike proteins”, given all that he and Shi Zhengli previously disclosed in their papers long before 2015.
Baric-1990
Just so you appreciate how long Ralph Baric has been at this game — he was designing recombinant coronaviruses way before there were any DNA sequencing machines or other modern tools of genetic engineering. Here is his paper on the creation of “temperature mutants” from mouse coronavirus from 1990:
The A59 strain of mouse hepatitis virus (MHV-A59) was used throughout the course of this study. Virus was propagated and cloned three times in the continuous murine astrocytoma cell line (DBT).
…
Various combinations of [temperature sensitive] mutants were mixed and inoculated onto cells at a multiplicity of infection of 10 each.
So Dr. Baric has been creating mutant viruses for over 30 years.
Wuhan-2017
The Shi Zhengli group has also not been idle since the famous 2015 paper. In 2017, they published a paper where they reported creating not one but 8 chimeric viruses — all made using transplanted RBDs from bat SARS-like viruses which they collected over a span of 5 years from the very cave around Kunming, Yunnan Province, where Shi Zhengli originally found Rs3367 and RsSCH014.
Using the reverse genetics technique we previously developed for WIV1 [23], we constructed a group of infectious bacterial artificial chromosome (BAC) clones with the backbone of WIV1 and variants of S genes from 8 different bat SARSr-CoVs. Only the infectious clones for Rs4231 and Rs7327 led to cytopathic effects in Vero E6 cells after transfection (S7 Fig). The other six strains with deletions in the RBD region, Rf4075, Rs4081, Rs4085, Rs4235, As6526 and Rp3 (S1 Fig) failed to be rescued, as no cytopathic effects was observed and viral replication cannot be detected by immunofluorescence assay in Vero E6 cells (S7 Fig). In contrast, when Vero E6 cells were respectively infected with the two successfully rescued chimeric SARSr-CoVs, WIV1-Rs4231S and WIV1-Rs7327S, and the newly isolated Rs4874, efficient virus replication was detected in all infections (Fig 7).

Full-length genome sequences of all SARSr-CoV detected in bats from the cave investigated in this study were used as reference sequences. The analysis was performed with the Kimura model, a window size of 1500 base pairs and a step size of 150 base pairs.
The authors then checked if their chimeras can infect human cells, and this time they used a live synthetic virus, rather than not pseudo-typed HIV constructs as before:
To assess whether the three novel SARSr-CoVs can use human ACE2 as a cellular entry receptor, we conducted virus infectivity studies using HeLa cells with or without the expression of human ACE2. All viruses replicated efficiently in the human ACE2-expressing cells. The results were further confirmed by quantification of viral RNA using real-time RT-PCR (Fig 8).
Baric-2019
Ralph Baric also showed no signs of slowing down. At the end of October 2019, his group submitted for publication another paper on the importance of spike protein protease cleavage (remember the furin site?) to crossing the “barrier to zoonotic infection” by coronaviruses:
Together, these results demonstrate that protease cleavage is also the primary barrier to infection of Vero cells with HKU5-CoV. Examining further, we compared the predicted cleavage at S1/S2 border, S2’, and the endosomal cysteine protease site across MERS, PDF2180, and HKU5 spikes (Fig. 6D) (26). For the S1/S2 site, MERS, Uganda, and HKU5 maintain the RXXR cleavage motif, although the different interior amino acids may alter efficiency. For the S2’ sequence, MERS and HKU5 also retain the RXXR motif; however, the Uganda spike lacks the first arginine (SNAR), potentially impacting cleavage.
As I recall the spirit of scientific competition between the groups of Baric and Shi Zhengli, I can’t help but wonder whether someone was conducting similar research in the Wuhan lab in 2019.
Gain-of-Function: Risky Business
Many people who first learn about the above research ask a very valid question: “But why?” Why do scientists create chimeric killer viruses? The politically correct answer is to develop preventive measures (drugs or vaccines) from possible natural chimeras and to understand the risks of their occurrence. Here, in fact, is what Baric, Shi Zhengli, and co-authors themselves wrote on this subject in their famous 2015 paper:
In addition to offering preparation against future emerging viruses, this approach must be considered in the context of the US government–mandated pause on gain-of-function (GOF) studies. On the basis of previous models of emergence (Fig. 4a,b), the creation of chimeric viruses such as SHC014-MA15 was not expected to increase pathogenicity. Although SHC014-MA15 is attenuated relative to its parental mouse-adapted SARS-CoV, similar studies examining the pathogenicity of CoVs with the wild-type Urbani spike within the MA15 backbone showed no weight loss in mice and reduced viral replication. Thus, relative to the Urbani spike–MA15 CoV, SHC014-MA15 shows a gain in pathogenesis (Fig. 1). On the basis of these findings, scientific review panels may deem similar studies building chimeric viruses based on circulating strains too risky to pursue, as increased pathogenicity in mammalian models cannot be excluded. Coupled with restrictions on mouse-adapted strains and the development of monoclonal antibodies using escape mutants, research into CoV emergence and therapeutic efficacy may be severely limited moving forward. Together, these data and restrictions represent a crossroads of GOF research concerns; the potential to prepare for and mitigate future outbreaks must be weighed against the risk of creating more dangerous pathogens. In developing policies moving forward, it is important to consider the value of the data generated by these studies and whether these types of chimeric virus studies warrant further investigation versus the inherent risks involved.
Were these words prophetic? At the end of 2014, the United States introduced a moratorium on state financing of such gain-of-function studies, but it was shortly canceled (in 2017). In China, no moratorium on such studies was introduced, on the contrary, they went full steam ahead with creating new “super labs” of the highest biosafety level (BSL-4), as in 2017 in Wuhan:

To be clear, the Wuhan lab was allowed to work with coronaviruses even before 2017, as these viruses only required a BSL-3 rating which the Wuhan Institute of Virology had. But their aspirations to obtain BSL-4 made a lot of people uneasy, including fellow researchers:
Future plans include studying the pathogen that causes SARS, which also doesn’t require a BSL-4 lab, before moving on to Ebola and the West African Lassa virus, which do. Some one million Chinese people work in Africa; the country needs to be ready for any eventuality, says Yuan. “Viruses don’t know borders.”
…
The plan to expand into a network heightens such concerns. One BSL-4 lab in Harbin is already awaiting accreditation; the next two are expected to be in Beijing and Kunming, the latter focused on using monkey models to study disease.Lina says that China’s size justifies this scale, and that the opportunity to combine BSL-4 research with an abundance of research monkeys — Chinese researchers face less red tape than those in the West when it comes to research on primates — could be powerful. “If you want to test vaccines or antivirals, you need a non-human primate model,” says Lina.
But Ebright is not convinced of the need for more than one BSL-4 lab in mainland China. He suspects that the expansion there is a reaction to the networks in the United States and Europe, which he says are also unwarranted. He adds that governments will assume that such excess capacity is for the potential development of bioweapons.
“These facilities are inherently dual use,” he says. The prospect of ramping up opportunities to inject monkeys with pathogens also worries, rather than excites, him: “They can run, they can scratch, they can bite.”
Trevan says China’s investment in a BSL-4 lab may, above all, be a way to prove to the world that the nation is competitive. “It is a big status symbol in biology,” he says, “whether it’s a need or not.”
Interestingly, in addition to Wuhan, the Chinese government planned to open a new BSL-4 lab in Kunming, with an eye to testing vaccines on primates. As you might recall, Kunming is not only the capital of Yunnan, but it is also where Shi Zhengli found the strains Rs3367 and RsSHC014 in nearby caves. By the way, primate testing was mentioned by Baric and Shi Zhengli as possible next steps for the development of preventive vaccines against potential future outbreaks of coronaviruses in their famous 2015 paper:
However, further testing in nonhuman primates is required to translate these finding into pathogenic potential in humans. Importantly, the failure of available therapeutics defines a critical need for further study and for the development of treatments. With this knowledge, surveillance programs, diagnostic reagents and effective treatments can be produced that are protective against the emergence of group 2b–specific CoVs, such as SHC014, and these can be applied to other CoV branches that maintain similarly heterogeneous pools.
Maybe by 2019 the creation and testing of potential vaccines against various SARS-like coronaviruses was already in full swing.
Beware of Lab
Let’s now take a look at the lab leak hypothesis. But first, I will provide some historical context, including previous confirmed lab leaks, as many of those happened before. First and foremost, lab leaks of the first SARS-CoV: initially, in the summer of 2003 in Singapore, then in December 2003 in Taiwan, and in the spring of 2004 twice in Beijing.
There were close calls in Europe and the USA, although thankfully no infections occurred there. For example, a French lab once lost vials with SARS, and an American BSL-4 laboratory in Texas, lost a vial containing Guanarito (Venezuelan hemorrhagic fever virus):
Only one scientist worked with the virus, and Reyes said the lab suspects that scientist accidentally threw the vial away in November.
…
Galveston biolab requires the most stringent safety measures because it studies biosafetly level BSL-4 materials, or dangerous infectious diseases that have no vaccines or cures. BSL-4 materials include Guanarito, Ebola and smallpox.
History knows other, much larger-scale leaks. For example, the “resurrection” of the H1N1 flu virus in 1977, which had previously been considered extinct. Yes, this is the virus of the famous “Spanish flu”:
Human influenza H1N1 viruses appeared with the 1918 pandemic, and persisted, slowing accumulating small changes in its genome (with a major change in 1947), until the H2N2 “Asian” flu appeared in 1957, causing a worldwide pandemic. H1N1 influenza virus then apparently became extinct, and was not isolated for 20 years. In 1969 the “Hong Kong” H3N2 virus replaced the H2N2 virus, and is still circulating.
In September 1977 an H1N1 influenza virus was isolated from human infections in the Far East region of the Soviet Union, and in early 1978 the Chinese reported they had isolated H1N1 virus in May of 1977 in northeast China adjacent to the Soviet outbreak. Using the early genetic tools available at the time, the 1977 H1N1 virus was found to be closely related to H1N1 human influenza viruses circulating in 1949–1950, but not to those circulating earlier or later.
…
Only since 2009–2010 did major papers begin to state directly the 1977 emergence of H1N1 influenza was a laboratory related release: “The most famous case of a released laboratory strain is the re-emergent H1N1 influenza A virus which was first observed in China in May of 1977 and in Russia shortly thereafter.”
…
The speculation that the 1977 release may have been related to H1N1 vaccine research is supported by the observation that in the initial outbreaks in China, nine of the ten viral isolates expressed “temperature sensitivity” (Kung 1978). Temperature sensitivity normally an uncommon trait, but one that was in the 1970s (and still is) a fundamental trait for making live attenuated influenza vaccines. Temperature sensitivity generally occurs only after a series of substantial laboratory manipulations and selections.Interestingly, further investigation indicated the circulating strains in 1977–78 were often comprised of mixed temperature-sensitive and normal components, and that temperature sensitivity apparently disappeared from the post-1978 H1N1 lineage rapidly. Escape of a mid-protocol population of H1N1 virus undergoing laboratory selection for temperature sensitive mutants would provide such a mixed population. In 1976–77 laboratory personnel in their late teens or early 20s would not have been exposed to pre-1957 H1N1 influenza viruses, and been susceptible to laboratory infections. The low severity of the 1977 pandemic might be in part due to the temperature sensitivity of the virus, a trait that limits virus replication in pulmonary tissues.
It seems that the creation of temperature-sensitive viral mutants to develop potentially attenuated vaccines was widespread at the end of the twentieth century. If you remember, in 1990, Ralph Baric himself also experimented with the creation of temperature-sensitive coronavirus strains.
Could something like this have caused the Covid-19 pandemic? Several options are possible — from a leak during development of a potential vaccine to fundamental research on laboratory recombination of the bat and pangolin viruses. Some particularly ambitious researcher could even decide to combine the two “fashionable research themes” — adding a furin site and transplanting RBM from a strain of one species (pangolin) to another (bats), so that later, confirming the increased virulence of the new chimeric virus, they can wax poetic about the dangers of the same recombination happening in Yunnan caves or wet markets. And if such a researcher could even pre-emptively develop a vaccine against this and other potential chimeras, all sorts of accolades could await.
Am I then saying this is what happened? Of course not, I do not claim to know what happened. Today, there is no evidence of this. For now, there is just a series of strange coincidences — for example, that the outbreak of the Yunnan coronavirus occurred thousands of kilometers from Yunnan in a wet market closest to the Wuhan Institute of Virology. Or maybe not at the wet market, as 3 of the first 4 patients had no ties to the market. Plus, there are coincidences in the structural features of the CoV2 genome, which resemble manipulations that virologists have repeatedly carried out in the lab. But coincidence is not proof.
Moreover, coincidences happen, and CoV2 could obviously have arisen naturally. It is not yet clear exactly how — for this, the bat and pangolin strains must have met in the same cell of some animal in Wuhan, since the outbreak occurred there (otherwise we would have seen other outbreaks along the path that animal would have taken to get to Wuhan). Given that bats were not sold in the Wuhan market, and generally hibernate at this time of the year, and that no other carriers of ancestral strains have yet been identified, the exact scenario of natural emergence remains a mystery.
On the opposite side of the balance, giving credence to the lab hypothesis, there are reports that in 2018, American experts were quite alarmed after their visit to the Wuhan Institute of Virology and conversation with Shi Zhengli. Their “lab tour” resulted in two diplomatic dispatches to Washington in which they noted a number of safety weaknesses:
Sources familiar with the cables said they were meant to sound an alarm about the grave safety concerns at the WIV lab, especially regarding its work with bat coronaviruses. The embassy officials were calling for more U.S. attention to this lab and more support for it, to help it fix its problems.
…
“During interactions with scientists at the WIV laboratory, they noted the new lab has a serious shortage of appropriately trained technicians and investigators needed to safely operate this high-containment laboratory,” states the Jan. 19, 2018, cable, which was drafted by two officials from the embassy’s environment, science and health sections who met with the WIV scientists. (The State Department declined to comment on this and other details of the story.)
The Chinese researchers at WIV were receiving assistance from the Galveston National Laboratory at the University of Texas Medical Branch and other U.S. organizations, but the Chinese requested additional help. The cables argued that the United States should give the Wuhan lab further support, mainly because its research on bat coronaviruses was important but also dangerous.
It is somewhat ironic the Wuhan lab received guidance from the Texas laboratory in Galveston, which at one time had itself lost a vial with a Guanarito virus: Wuhan specialists were trained at Galveston, which was even reported in the Wuhan Institute’s own newsletter (though, that publication has been deleted from the WIV website, but it is still available at the Wayback Machine):

A couple of final touches to the family portrait of laboratory leaks: in November 2019, an outbreak of brucellosis (a bacterial infection) occurred in two research centers in Lanzhou, China, infecting over 100 researchers who worked there. American labs have also not been immune to outbreaks, although not on the same scale:
Possible Hallmarks of Lab Origin?
Let us now turn our attention back to the virus itself. Does it have any obvious signs of lab manipulation? First, a few words about what “obvious” means. Any mutation can arise naturally, and even if the amino acid insert that had created the furin site in CoV2 was not “PRRA” but “MADEINWVHANPRRA”, there would still be a non-zero chance that it arose by accident. But for us, and for any court, I think this would be enough to prove lab origin beyond a reasonable doubt.
The main problem with such evidence is that even in a lab-made virus it simply may not exist. Basically, a good genetic engineer can create a synthetic virus that would be indistinguishable from a natural one. Moreover, often researchers deliberately introduce some synonymous mutations into their designs so that later they can discern their strain from natural ones. But if the creators choose not to reveal these markers, it is impossible to distinguish them from natural mutations.
But sometimes traces of manipulation may remain, especially if the creators do not try to hide them. First of all, I am talking about the spots in virus genome where its DNA is cut (recall that RNA virus manipulations are carried out in complementary DNA constructs). This occurs when virus creators need cut out a segment, or stitch together new segments. After all, DNA cannot be cut in arbitrary places (CRISPR aside), but only where its nucleotide sequence (usually 4–6 “letters”) forms a sequence recognized by some restriction enzyme, that is, an enzyme that can cut a nucleotide chain. However, such an analysis is complicated by the fact that there are hundreds of different types of restriction enzymes used in genetic engineering. But let’s try it anyways.
As a baseline, here is an example of the work of the Baric group from 2008, where they took Bat-SCoV and replaced its RBD by an RBD from human SARS. Here’s how they describe the creation of their chimera:
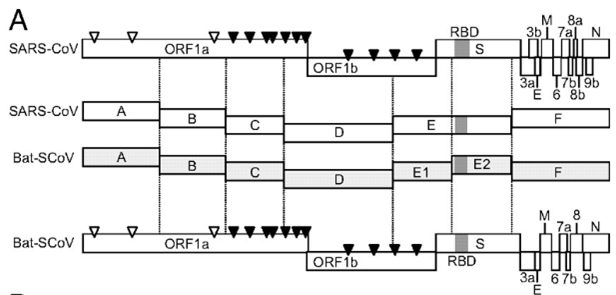
(A) Schematic representation of SARS-CoV and Bat-SCoV (GenBank accession no. FJ211859) genomes and reverse genetics system. (Top) Arrowheads indicate nsp processing sites within the ORF1ab polyprotein (open arrowheads, papain-like proteinase mediated; filled arrowheads, nsp5 [3C-like proteinase] mediated). Immediately below are the fragments used in the reverse genetics system, labeled A through F. The fragments synthesized to generate Bat-SCoV exactly recapitulate the fragment junctions of SARS-CoV with the exception that the Bat-SCoV has 2 fragments, Bat-E1 and Bat-E2, which correspond to the SARS-E fragment.
As you can see, the Baric group first created a synthetic clone of Bat-SCoV in the same pattern as they used for their synthetic clone of SARS-CoV. That is, for the bat clone, they used the same 6 segments with the same restriction enzyme sites that they had previously used for SARS-CoV, which allowed them to swap virus segments between different strains like Lego pieces. Here is their detailed description:
Viruses containing PCR-generated insertions within the viral coding sequence were produced by using the SARS-CoV assembly strategy (24, 33, 53) with the following modifications. Briefly, for Bat-F virus, full-length cDNA was constructed by ligating restriction products from SARS-CoV fragments A–E and Bat-SCoV fragment F, which required a BglI-NotI digestion. For Bat-SCoV and Bat-SRBD, Bat-SRBM, and Bat-Hinge, plasmids containing the 7 cDNA fragments of the Bat-SCoV genome were digested by using BglI for Bat-A, Bat-B, Bat-C, and Bat-D, BglI and AflII for Bat-E1 and Bat-E2, and BglI and NotI for Bat-F. Digested, gel-purified fragments were simultaneously ligated together. Transcription was driven by using a T7 mMessage mMachine kit (Ambion), and RNA was electroporated into Vero cells (24, 53).
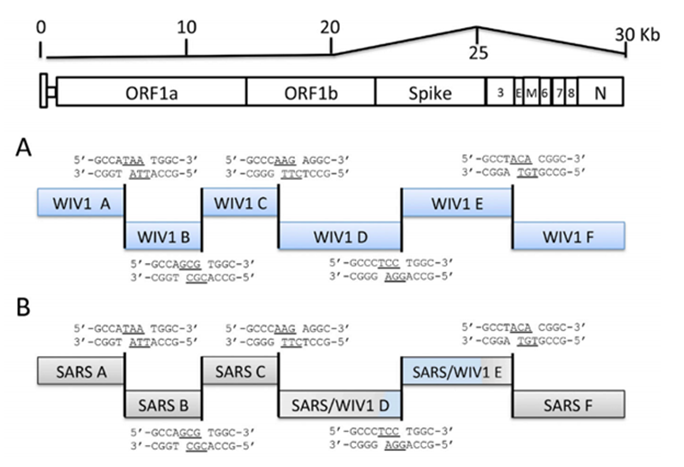
All these three-letter abbreviations (BglI, AflII, NotI, etc.) in the sentence highlighted above are different types of restriction enzymes. Let’s see if there are any differences in the restriction enzyme sites in the spike protein sequence of the chimera compared to the genome of the original SARS-CoV:
As can be seen, the restriction enzyme sites of the chimera are almost identical to those in the original sequences in Bat-SCoV or SARS from where they were taken. The only differences are noticeable at the “stitching” sites of the inserted SARS piece. Here, for example, is the left (5’-) edge of the insert:

Here Bat-SCoV and SARS turned out to have a common identical region of nucleotides (the intersection of cyan and pink regions), and there are no new restriction enzyme sites at the stitching site of the two sequences, on the contrary, the SspI site from SARS disappeared. And here is the right (3’-) edge of the insert:
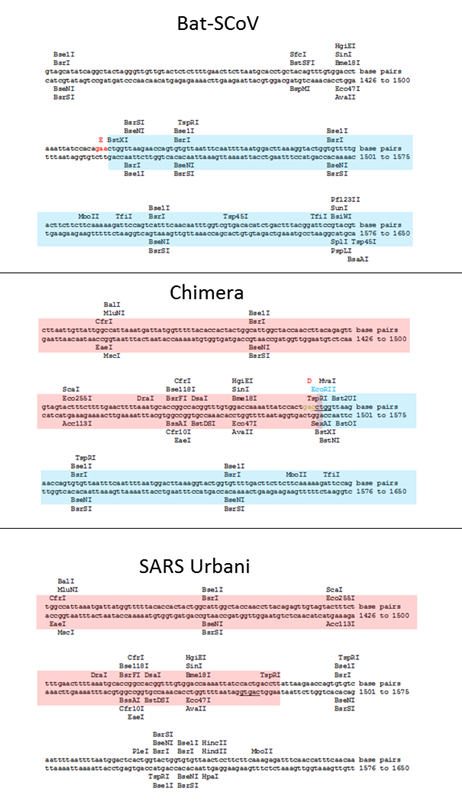
Here, on the contrary, all the original restriction enzyme sites remain at the site of ligation, and even new ones appear, for example, EcoRII. Had I not known that the chimeric genome is the result of lab manipulations, could I deduce this by looking at these 3 sequences? Not really, and even if some suspicion did creep in, it would certainly not be beyond a reasonable doubt. Perhaps it would be obvious to specialists in genetic engineering by some other signs, and, if so, I hope they speak up.
But in any case, let’s compare the RaTG13spike protein to CoV2 and pangolin-2019. Just in case something does jump out.
This is what the RBD (highlighted in light green) and RBM (yellow) look like for all three:
So, anything interesting? Well, I noticed some new restriction enzyme sites in CoV2 marked by red rectangles — they coincide with unique mutations in the amino acid sequence (also marked by red rectangles in the amino acid sequences on the far right). Just in case, I highlighted several other new sites: blue rectangles, and a green rectangle located in the region of the only amino acid that differs between RBMs of CoV2 and pangolin-2019.
Let’s now compare the stretch around the PRRA insert that created the furin site in CoV2 among those three strains:

Here, too, several new restriction sites have appeared (highlighted in blue) on both sides of the new insert. Could they have been used to create a furin site? Theoretically, yes. Alternatively, the insertion could have been made via existing sites or even using the “seamless” ligation method — i.e. by creation of segments with new restriction sites which disappear after the complementary ends are joined. You might remember that the Baric group have applied this technology in 2002 to create a synthetic clone of murine coronavirus:
The interconnecting restriction site junctions that are located at the ends of each cDNA are systematically removed during the assembly of the complete full-length cDNA product, allowing reassembly without the introduction of nucleotide changes.
In 2003 they have used this approach again for a synthetic clone of SARS-CoV:
To rapidly assemble consensus clones, we used class IIS restriction endonucleases that cut at asymmetric sites and leave asymmetric ends. These enzymes generate strand-specific unique overhangs that allow the seamless ligation of two cDNAs with the concomitant loss of the restriction site.
Today, genetic manipulation techniques are so advanced and have become so routine that the October 2019 Beijing paper which had inserted a new furin site into the chicken coronavirus, only devoted a couple of sentences to their methodology:
2.2. Generation of Recombinant Virus
Recombinant rYN-S2/RRKR virus containing an S protein with the furin-S2′ site was generated by vaccinia recombination, as described previously [20,28]. Briefly, plasmid with the furin-S2′ site was generated using the Seamless Assembly kit (Invitrogen, Carlsbad, CA, USA) and transfected into CV-1 cells infected by vaccinia virus containing the genome of YN-ΔS-GPT. Furin-S2’ site was introduced into the YN cDNA by homologous recombination using the transient dominant selection system [25].
The pace of progress in genetic engineering is astounding. Here is a description of the above Seamless Assembly kit:
The GeneArt® Seamless Cloning and Assembly Kit enables the simultaneous and directional cloning of 1 to 4 PCR fragments, consisting of any sequence, into any linearized vector, in a single 30-minute room temperature reaction. The kit contains everything required for the assembly of DNA fragments, and their transformation into E. coli for selection and growth of recombinant vectors.
• Speed and Ease — Clone up to 4 DNA fragments, with sequence of your choice, simultaneously in a single vector (up to 13 Kb); no restriction digestion, ligation or recombination sites required
• Precision and Efficiency — Designed to let you clone what you want, where you want, in the orientation you want, and achieve up to 90% correct clones with no extra sequences left behind
• Vector Flexibility — Use our linear vector or a vector of your choice
• Free Tools — Design DNA oligos and more with our free web-based interface that walks you step-by-step through your project
• Diverse Applications — Streamline many synthetic biology and molecular biology techniques through the rapid combination, addition, deletion, or exchange of DNA segments
Up to 4 DNA fragments can be joined in a desired orientation in about half an hour, without having to deal with restriction enzymes or ligation. Once you’re done, quickly “upload” your creation into E. coli to propagate the resulting design. Easy-peasy!
In summary, the restriction enzyme site analysis did not yield anything conclusive. It did, however, point out that not only CoV2 is quite unique, but so is RaTG13, and we should continue digging into the origins of both.
Codon Preferences
For these purposes, I decided to take a look at codon usage bias to check which strains look like CoV2 and RaTG13 the most. It is known that viruses tend to adapt their codon signature to the preferences of their hosts, so I expected to see RaTG13 exhibit a similar pattern to other bat viruses, and also hoped to see a difference from pangolin strains.
SARS-CoV, for example, is very similar to Rs3367 and RsSCH014, as one might expect:
Among themselves, by the way, SARS, MERS and CoV2 do differ:
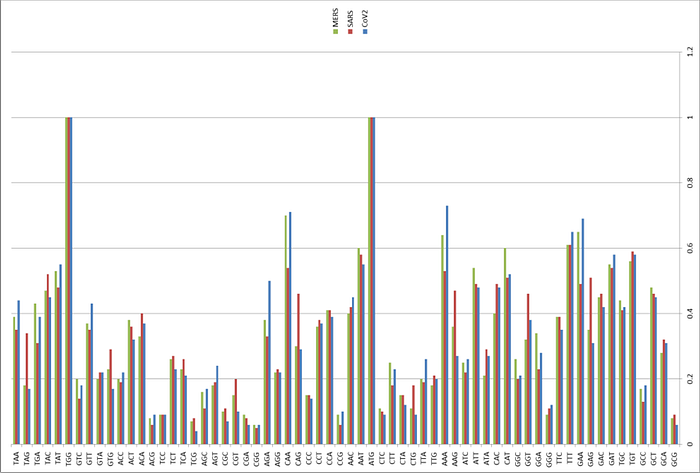
RaTG13 is similar to CoV2, which is also to be expected:
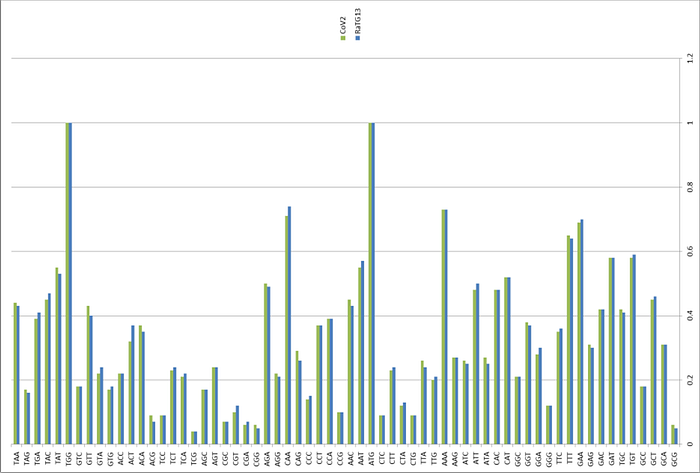
But RaTG13 is actually not that close to the pangolin strains, and the pangolin strains are not exactly identical to each other:
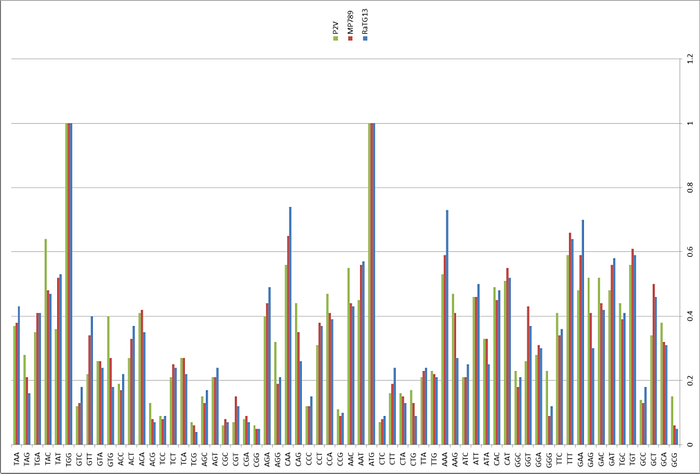
RaTG13 also differs from ZXC21 and ZC45:
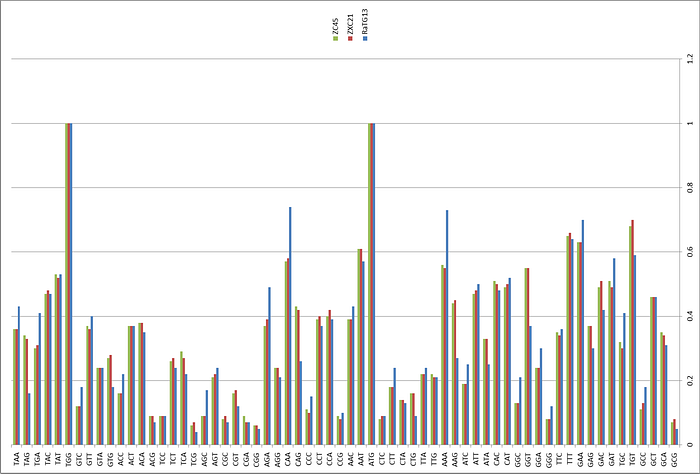
Looking at Yunnan strains, RaTG13 is quite distant from Rs3367 and RsSCH014, and closer to LYRa11, but also with noticeable differences:
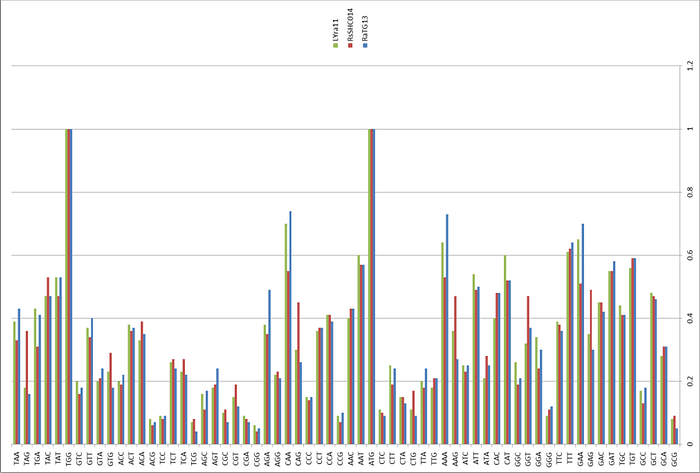
In general, as before, RaTG13 and CoV2 stand out in a class of their own. I was also intrigued by the AAA codon — they use it much more often than their fellow strains:
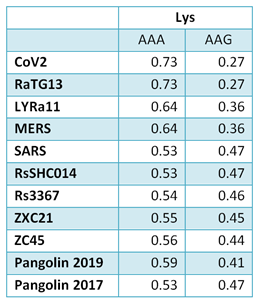
This is probably just another coincidence, but a similar proportion between AAA and AAG is observed in E. coli. Can the cDNA codon signature change if it is being cultivated for a long time in cell culture? Maybe, but I haven’t yet dug into this topic very deeply.
[UPDATED] I also decided to check codon usage patterns between RaTG13 and other Ra strains collected from the same abandoned mineshaft in Mojiang where in 2013 Shi Zhengli’s group found strain RaBtCoV/4991 (KP876546) that shares an identical 370-bp RdRp segment with RaTG13. Unfortunately, only 816-bp segments of the RdRp gene were available for the other Ra strains (RaBtCoV/3750 and RaBtCoV/4307–2), so I extracted the corresponding 816-bp segment from RaTG13 for the purposes of codon usage comparison. RaTG13 again differed substantially, while the other two strains clustered together:
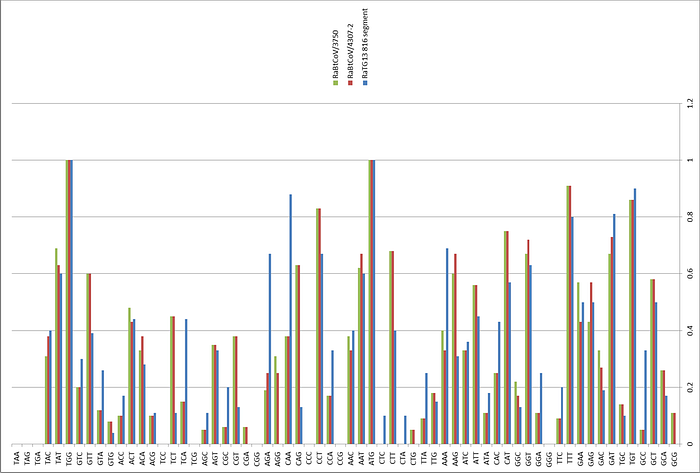
So codon analysis also did not reveal any obvious signs of lab origins, but once again confirmed the uniqueness of CoV2 and RaTG13. What does this leave us with? So far, just a number of oddities, which, as scientists like to say, taken together, do not allow us to reject the lab origin hypothesis of CoV2.
The Nature Paper vs. the Lab-Made Hypothesis
But didn’t that Nature article refute the lab-made hypothesis? No, not really. There is no irrefutable evidence against it in the paper, just a loud “we don’t believe so” based on a shaky foundation. Judge for yourself — here are the authors’ key arguments in support of their conclusions:
While the analyses above suggest that SARS-CoV-2 may bind human ACE2 with high affinity, computational analyses predict that the interaction is not ideal and that the RBD sequence is different from those shown in SARS-CoV to be optimal for receptor binding. Thus, the high-affinity binding of the SARS-CoV-2 spike protein to human ACE2 is most likely the result of natural selection on a human or human-like ACE2 that permits another optimal binding solution to arise. This is strong evidence that SARS-CoV-2 is not the product of purposeful manipulation.
In the original paper, the quoted sentences are just below the diagram showing identical RBMs between CoV2 and pangolin-2019. So I am puzzled as to what “computational analysis” has to do with anything. Obviously, the most likely scenario for the lab-made hypothesis is the transfer of RBM from one strain to another — which virologists have done many times before. Therefore, the author’s chain of arguments does not make sense: “computer says binding is not ideal, thus CoV2 must be the result of natural selection. Ergo, this is strong evidence that CoV2 is not lab-made.” Wait, just because CoV2 differs from some “optimal” virus, doesn’t mean it could not have been created in a lab. Not the lab trying to create “optimal” bioweapons, but a lab creating chimeras of naturally found strains, say, in bats and pangolins.
The authors continue to surprise:
Furthermore, if genetic manipulation had been performed, one of the several reverse-genetic systems available for betacoronaviruses would probably have been used. However, the genetic data irrefutably show that SARS-CoV-2 is not derived from any previously used virus backbone.
Again, the same questionable logic dressed in categorical adjectives: “genetic analysis irrefutably proves that CoV2 was not created on the basis of previously known strains!” Well thanks, Captain Obvious. But why couldn’t potential creators of CoV2 make a cDNA backbone from unpublished strains related to or even derived from RaTG13? Then they could easily insert the pangolin RBM into it, as well as add a furin site (or maybe the cDNA backbone already had one). Virologists have been doing things like this for 20 years, and modern genetic engineering tools make such manipulations accessible even to a grad student.
As for the chances of the furin site arising in cell culture, the authors also express strange ideas:
The acquisition of both the polybasic cleavage site and predicted O-linked glycans also argues against culture-based scenarios. New polybasic cleavage sites have been observed only after prolonged passage of low-pathogenicity avian influenza virus in vitro or in vivo. Furthermore, a hypothetical generation of SARS-CoV-2 by cell culture or animal passage would have required prior isolation of a progenitor virus with very high genetic similarity, which has not been described. Subsequent generation of a polybasic cleavage site would have then required repeated passage in cell culture or animals with ACE2 receptors similar to those of humans, but such work has also not previously been described.
First off, the authors themselves cite previous works where the furin site arose in vitro as viruses were cultured in cells. And second, what do they mean, a strain with high genetic similarity has not been described — what about RaTG13? If it had its RBM replaced by one from the pangolin strain, and then the chimeric strain was cultured in vitro, then the furin site could well have arisen in this matter. Additionally, the new strain could thus acquire other mutations that distinguish CoV2 from RaTG13 and pangolin-2019.
But in terms of the potential lab-based origin of the furin site, I am more inclined to hypothesize a specific insertion — as in the Beijing paper from October 2019 with chicken coronavirus. After that, the synthetic strain could have acquired new mutations by subsequent culturing in vitro or in vivo — like the MA15 murine strain in 2007, for example. Or maybe even using the same mouse model with humanized lung tissues and immune system that was created at UNC by Baric’s and other groups in 2018, in which they reported testing several viruses including MERS:
The human innate and adaptive immune system of BLT-L mice
We generated an in vivo model with human lung implants and an autologous human immune system by constructing BLT mice with autologous human lung implants (BLT-L humanized mice).
Finally, even if CoV2 is the product of selection rather than intelligent design, that does not rule out a lab leak either — selection can happen in the lab just as well, both natural and artificial kinds. Different strains can recombine in research animals or in vitro by design or by chance.
On the 4% Genome Difference between RaTG13 and Cov2
Some critics of the lab-made hypothesis claim that the observed ~4% genetic difference between RaTG13 and CoV2 is too high to have possibly occurred in a lab if RaTG13 itself was used as a backbone. Observed mutation rates for RNA viruses vary widely — from 10⁻⁶ to 10⁻⁴ nucleotides per replication in vitro, and in humans CoV2 seems to mutate at a rate of 25 mutations per year. Thus, the logic goes, it would take years, if not decades, for two strains to diverge by 4%. While that is a valid point, there are several issues with that line of reasoning.
First, in vitro mutation speeds (i.e. per unit of time) are much higher, as you can passage cells much more often than infect new animals. As SARS and MERS in vitro experiments showed, significant mutations might be observed after only a few passages. For example, the 2004 paper reported that only after 600 passages there already was a 2.1% difference in the genomic sequences of spike proteins between the original strain and its progeny:
Moreover, in the presence of some antiviral compounds, such as nucleoside analogs (e.g. ribavirin or remdesivir), mutation rates in RNA viruses can increase even further:
We obtained an estimate of the spontaneous mutation rate of ca. 10⁻⁴ substitutions per site or lower, a value within the typically accepted range for RNA viruses. A roughly threefold increase in mutation rate and a significant shift in mutation spectrum were observed in samples from patients undergoing 6 months of interferon plus ribavirin treatment. This result is consistent with the known in vitro mutagenic effect of ribavirin and suggests that the antiviral effect of ribavirin plus interferon treatment is at least partly exerted through lethal mutagenesis.
So if ancestral CoV2 was being lab-tested to assess how its mutagenesis might affect the efficacy of potential vaccines or antiviral drugs, it could have accumulated mutations at a much higher rate.
But possibly, the biggest problem with the 4% difference argument is that it relies on RaTG13 being exactly what WIV says it is. If we are to seriously consider the lab leak hypothesis, we must concede that it does not make sense to blindly trust the data released by the very lab suspected of the leak. If the leak did occur, as is the premise of the lab hypothesis, then the description of what RaTG13 is could be furthering the goal of covering up the leak.
Again, I am not claiming with certainty that is what is happening here. All I am saying is that this is what could have happened, and we need a lot more evidence before we can reach a definitive conclusion. One thing that could help rule out tampering with RaTG13 is having independent labs sequence the 2013 Yunnan samples that She Zhengli extracted RaTG13 from. WIV must still have them if they re-sequenced RaTG13 in 2020.
Shi Zhengli-2020
As I was writing this post, a fresh paper co-authored by Shi Zhengli came out, in which the authors tested a peptide which they have been studying for some time before against CoV2. That peptide was meant to be a pan-coronavirus inhibitor, and its designed mode of action was to block the fusion of a spike protein with a cell membrane. The authors, of course, mention the new furin site of CoV2, and suggest that it may play an important role in the much more efficient penetration of CoV2 into the cell:
In this study, we have shown that SARS-CoV-2 exhibits much higher capacity of membrane fusion than SARS-CoV, suggesting that the fusion machinery of SARS-CoV-2 is an important target for development of coronavirus fusion inhibitors.
…
Generally, β-B coronaviruses lack the S1/S2 furin-recognition site, and their S proteins are uncleaved in the native state. For example, SARS-CoV enters into the cell mainly via the endosomal membrane fusion pathway where its S protein is cleaved by endosomal cathepsin L and activated. Inducing the S1/S2 furin-recognition site could significantly increase the capacity of SARS-CoV S protein to mediate cellular membrane surface infection.
In this context, I wonder whether the authors have previously conducted experiments on how adding a furin site can alter the effectiveness of their peptide (or other drugs or vaccines) against a given coronavirus.
Not to be outdone, Ralph Baric also joined the race to find drugs against CoV2. As I understand, he and co-authors took data on the effectiveness of their nucleoside analogue (β-D-N4-hydroxycytidine, NHC) against SARS-CoV and MERS that they already had, added some in vitro data on CoV2, and sent off the paper to print. Nucleoside analogues (such as the famous remdesivir) are a fundamentally different approach than Shi Zhengli et al. Here, the authors try to prevent viral replication by giving “defective” letters of the genetic alphabet to virus’ copying machine, while Shi Zhengli and coauthors try to prevent the virus from entering the cell altogether. Theoretically, these approaches could be combined.
This is the End, Beautiful Friend
If you made it here by reading rather than scrolling, mad props to you. Hey, even if you scrolled, that’s cool too, and I apologize for the verbosity. I just didn’t anticipate that the rabbit hole would turn out to be a whole underground cave system. I hope that you found this deep dive into the world of virology interesting and enjoyed the exploration of the lab-made CoV2 hypothesis. In my opinion, the data I have presented, taken together, do not allow us to reject this possibility.
Let me be clear: this does NOT prove that CoV2 was synthesized in the laboratory. Yes, as we have seen above, from a technical standpoint, it would not be difficult for a modern virologist to create such a strain. But there is no direct evidence that anyone did this, and strange coincidences cannot pass for circumstantial evidence. On balance, the current chances against this are still higher than for the natural origins of CoV2. Moreover, even if CoV2 was indeed an unfortunate lab leak, the scientists themselves are not to blame, as they were working within the established international laws and guidelines on such research. Now, those who might be trying to cover up that leak, that’s a different story.
The opposite point is worth repeating too: the inverse hypothesis about the exclusively natural origin of the virus does not yet have strong evidence either. Until intermediate ancestors between RaTG13, pangolin-2019 and CoV2 are found, in whom we could trace the mosaic recombination that we observe in CoV2, the question of its origins remains open. In closing, there is no one better to quote on this matter than Ralph Baric himself:
What is the reservoir species of SARS-CoV-2?
They have not identified the actual reservoir species. Reports show that pangolins are potentially the intermediate host, but pangolin viruses are 88–98% identical to SARS-CoV-2. In comparison, civet and racoon dog strains of SARS coronaviruses were 99.8% identical to SARS-CoV from 2003. In other words, we are talking about a handful of mutations between civet strains, racoon dog strains and human strains in 2003. Pangolins [strains of CoV2] have over 3000 nucleotide changes, no way they are the reservoir species. Absolutely no chance.
So there you have it. It remains possible that the mysterious virus host was a lab:

Bad pun? Sorry, last one.
How I Learned to Hate the GOF
I hope this post is not used to prematurely assign blame or propagate one-sided theories. What I do hope it highlights is the scale of dangerous gain-of-function research that has been and is going on in virology. The Covid-19 pandemic really exposed its huge risks in the face of few benefits: GOF research hasn’t protected us from this outbreak, hasn’t provided us with any effective treatments or vaccines in time to save hundreds of thousands of lives lost to CoV2, and if there is even a 0.1% chance GOF research caused the whole thing, that chance is too high.