SARS-CoV-2 is precisely the virus WIV was hunting for in 2019

Lab leak critics often say that before the pandemic, the Wuhan Institute of Virology would not have considered a SARS2 progenitor interesting. “WIV was only interested in SARS-like viruses that were very close to SARS1,” they say.
In actuality, the exact opposite is true — in late 2018, EcoHealth and WIV outlined their updated virus hunting criteria for 2019–2023, which show that they were now looking for SARS-like viruses that were 10–25% different from SARS1 in their spike but could still enter human cells. SARS2 fits those criteria like a glove: its spike is 24% different from that of SARS1, and yet it binds to the human ACE2 receptor even better than SARS1.
Moreover, as I will show below, since SARS2 or its BANAL-like progenitor would have been able to evade SARS1-based vaccines and antibodies, such strains would have been prioritized and likely turned into full-genome synthetic backbones.
The good old days of 2018
Let’s travel back in time to November 2018. By this time, EcoHealth has been working closely with WIV for many years on a number of projects involving MERS-like and SARS-like viruses. On the SARS-like front, they had a 5-year NIH grant R01AI110964 (2014–2019) that was up for renewal at the time.
Just a few months prior, in March 2018, EcoHealth pitched a $14M mega-grant proposal for DARPA suggesting to sample thousands of bats in three Yunnan caves in search of novel SARS-like viruses, which the grant proposal authors then promised to “DEFUSE” by immunizing bats against them with “novel chimeric polyvalent recombinant spike proteins” (emphasis added):
Our goal is to defuse the potential for spillover of novel bat-origin high-zoonotic risk SARS-related coronaviruses in Asia. In TA1 we will intensively sample bats at our field sites where we have identified high spillover risk SARSr-CoVs. We will sequence their spike proteins, reverse engineer them to conduct binding assays, and insert them into bat SARSr-CoV (WIV1, SHC014) backbones (these use bat-SARSr-CoV backbones, not SARS-CoV, and are exempt from dual-use and gain of function concerns) to infect humanized mice and assess capacity to cause SARS-like disease.
…
In TA2, we will evaluate two approaches to reduce SARSr-CoV shedding in cave bats: (1) Broadscale immune boosting, in which we will inoculate bats with immune modulators to upregulate their innate immune response and downregulate viral replication; (2) Targeted immune boosting, in which we will inoculate bats with novel chimeric polyvalent recombinant spike proteins plus the immune modulator to enhance innate immunity against specific, high-risk viruses. … The most effective biologicals will be trialed in our test cave sites in Yunnan Province, with reduction in viral shedding as proof-of-concept.
As the DEFUSE grant proposal was not funded, parts of it made it to EcoHealth’s November 2018 grant renewal application for their NIH grant R01AI110964. Now EcoHealth and WIV were proposing to sample 5,000 bats in an attempt to “almost fully characterize the expected natural diversity of SARSr- and other β-CoVs in the region”. Their new focus on a particular subset of SARS-like strains was clear:
1.4.b Viral strain prioritization: Of the expected 100–200 novel SARSr-CoV strains, we will down-select to prioritize for further characterization based on S genes that are: i) different from SHC014, WIV1, SARS-CoV with diversity ranges of 10–25%; ii) have virus S RBD that could use human/bat receptors; iii) have recombinant chimeric spikes indicative of gene flow between clade I and II strains; iv) have bat ACE2 receptors that might select for spike RBDs that can use human receptors for entry (15/18 conserved residues in human/bat ACE2 molecules that bind SARSs-CoV S RBD domains are likely more efficient receptors than 3/18 conserved sites).
SARS2’s spike is 23% different from those of SCH014 or WIV1, and 24% different from that of SARS1. Similarly, the spikes of BANAL-52/103/236 strains are all 22% different from those of SCH014 or WIV1, and 23% different from SARS1's. But in terms of the ability to bind to the human ACE2 receptor, the RBD of SARS2-like strains is even better than SARS1, which would have been apparent even from computer modeling.
Here is what the grant authors planned to do with these strains (emphasis added):
We will use S protein sequences to select a range of viral strains that cover the 10-
25% S protein divergence we predict as high public health potential and construct chimeric SARSr-CoVs using the WIV1 backbone and these S genes as done previously (12, 18, 38). We will rescue of full-length clones and assess infection of non-permissive cells expressing human, bat and civet ACE2 receptors, Vero cells, primary human airway epithelial cells, and CaCo cells for HKU3r-CoVs (which have not been cultured and may use intestinal epithelium in nature). We will conduct experimental infections in hACE2 transgenic mice to
assess pathogenicity and clinical signs (18). Finally, using a panel of mAbs that neutralize SARS-CoV infection in vitro and in vivo, and vaccine against SARS-CoV S protein, we will examine the capacity of strains with divergent S protein sequences to evade therapeutics, revealing strains with high public health potential.
The last bit is particularly interesting. The grant renewal application elaborates (emphasis added):
3.3.b Primary human airway epithelial cell culture: …SARSr-CoVs that differ significantly in S protein sequence (11–24%) from epidemic SARS-CoV yet replicate in vitro, will also be evaluated for sensitivity to neutralization in Vero cells by PRNT50 assays using broadly SARS-CoV cross reactive human mAbs S227.14, S230.15, S215.17, and S109.8 (49, 55).
…
3.3.c Humanized mouse infection experiments: …hACE2 transgenic mice will be injected with SARS-CoV mAbs, and infected with chimeric bat SARSr-CoVs. Clinical signs and morbidity will be assessed and tissue pathology examined and compared with mice without treatment of mAbs to determine the therapeutic effect on SARSr-CoV infection, and protection of SARSr-CoV by wildtype SARS-S based vaccines assessed as described (56, 66). We will sequence full length genomes of high risk strains that are antigenically distinct and escape SARS cross neutralization, synthetically reconstruct a small subset (1–2) and evaluate the ability of nucleoside analogues to inhibit growth in HAE cultures and/or in vivo (55, 56).
The last sentence shows just how interested the grant authors were in novel SARS-like viruses that could escape SARS1-based neutralization. So interested that they planned to create synthetic backbones out of them for further testing in cells and/or mice to see if nucleoside analogs can inhibit their growth.
Previously, EcoHealth pitched this type of work to DARPA in their DEFUSE mega-grant (while promising WIV that they too will get to do some of this work if the grant gets funded, despite the proposal saying that this work was to be done at UNC):

Note that some nucleoside analogs (e.g. ribavirin or molnupiravir) are mutagens which are meant to inhibit viral growth by introducing a lot of mutations. The flip side of this, as the molnupiravir saga shows, is that mutagens can also accelerate viral evolution. When such mutagens are used during viral passaging in the lab (in vitro or in vivo), this could phylogenetically “age” passaged strains at a much quicker pace than what would have been observed in nature.
Escape from SARS1-based immunity
So why do I say that SARS2 or its BANAL-like progenitors would have grabbed the grant authors’ attention? Because not only do they fit the “10–25% spike difference while still binding to human ACE2" criteria, but they are also antigenically very distinct from SARS1 and are practically certain to evade SARS1-based mAbs or vaccines. Here’s why.
The “broadly SARS-CoV cross reactive human mAbs S227.14, S230.15, S215.17, and S109.8” that the grant renewal application names as the neutralization panel to test novel SARS-like CoVs against are the key to understanding this. In 2008, Ralph Baric’s group investigated these mAbs and has produced the following figure nicely summarizing which epitopes they target on the SARS1 spike protein:
Now, if we compare the spikes of SARS2 and its related strains to those of SARS1 in the above figure, we see something quite striking:
Namely, all of the epitopes in the receptor binding domain (RBD) and almost all other epitopes in S1 that Baric’s group has identified as being the targets of those mAbs are different in SARS2-like viruses.
Moreover, in 2010, the Baric group followed up on this work by creating SARS1 escape mutants against these and other mAbs. They’ve observed the following escape mutations in the RBD:
Again, we see that SARS2-like strains already have different amino acid residues than SARS1 in all but one location relevant for these mAbs. Taken together, these observations suggest those antibodies would almost certainly be ineffective against SARS2-like strains. Coupled with how well SARS2 or BANAL-52/236/103 bind to the human ACE2 receptor, this would have made such strains the top priority for the grant authors as per their November 2018 proposal.
Could the unique SARS2 R346 mutation be an escape mutation?
As an aside, I was intrigued to see that one of the SARS1 mAb S109.8 escape mutants from the 2008 Baric group paper had a K333N mutation in the RBD:
Four out of six plaques of the S109.8 escape mutants contained a single amino acid change at T332I, while two plaques contained a single amino acid change in an adjacent residue at position K333N.
That SARS1 escape mutation corresponds to a notable SARS2 RBD mutation (R346) relative to its ancestral bat strains. Moreover, that SARS2 location is the most frequent reversion (R346T) — i.e. mutation back to the amino acid residue present in ancestral strains — observed in over 11% of circulating SARS2 strains:
Based on this, I wonder whether the SARS2 R346 mutation could be a sign of SARS2 having encountered selective pressure similar to that exerted by the S109.8 antibody on the SARS1 escape mutant in the 2008 Baric group paper, because in 2016, the Baric group — in the paper titled “SARS-like WIV1-CoV poised for human emergence” — subjected WIV1 to neutralization testing with several antibodies, including S109.8, and then talked about the very same AA position 333 of SARS1/WIV1 spikes: the authors noted the different AA residues between SARS1 and WIV1 as the likely reason for reduced neutralization efficiency of S109.8 against WIV1 relative to SARS1 (emphasis added):
Having established a potential threat based on replication in primary human cells and preference for the human ACE2 receptor in vivo, we next sought to determine if monoclonal antibody therapies could be used to lessen disease similar to ZMApp for Ebola (13). We first tested a SARS-CoV monoclonal derived via phage display and antibody escape (Fm6) (14) and found both wild-type SARS-CoV Urbani and WIV1-MA15 were strongly neutralized at low antibody concentrations (Fig. 4A). Similarly, a panel of monoclonal antibodies derived from B cells from SARS-CoV–infected patients also prevented virus infection via WIV1-CoV spike (15, 16). Both antibodies 230.15 and 227.14 robustly inhibited WIV1-MA15 replication with kinetics similar to or exceeding SARS-CoV Urbani (Fig. 4 B and C). In contrast, antibody 109.8, which maps outside the receptor binding domain, produced only marginal neutralization of WIV1-MA15 (Fig. 4D). Whereas the residue associated with prior escape mutants was conserved at position 332, the adjacent residue had a significant change (K33[3]T) [332 instead of 333 was a typo in the paper — Y.D.] in WIV1-CoV, possibly contributing to reduced efficacy of this antibody.
S109.8 is even more relevant in the context of SARS2 because, as I have shown above, SARS2 has a very different RBD neutralization profile than SARS1 and thus antibodies like S230.15 and S227.14 that are very good at neutralizing SARS1 and SARS1-like strains (like WIV1) would almost certainly be ineffective against SARS2. But S109.8, on the other hand, “which maps outside the receptor binding domain”, as the Baric group noted, could still provide at least some “marginal neutralization” as it did for WIV1. After all, WIV1 and SARS2-like strains have identical residues in positions 332 and 333 (T and T; numbering is relative to the SARS1 spike). SARS2 itself, however, is unique among SARS2-like strains in having an R in that position (346 relative to SARS2 numbering), reminiscent of the 2008 S109.8 SARS1 escape mutants.
Was this work already underway in late 2019?
Now, fast forward a year to November 2019, and Peter Daszak, EcoHealth’s President, implies that some of the work described in their grant renewal application has already been done:
A few weeks later, in a December 9, 2019 interview, he repeated this claim:
“we’ve now found — after, you know, six or seven years of doing this — over a hundred new SARS-related coronaviruses very close to SARS. Some of them get into human cells in the lab, some of them can cause SARS disease in humanized mouse models and are untreatable with therapeutic monoclonals and you can’t vaccinate against them with the vaccine”
Interestingly, in the above interview Daszak mentions having found over a hundred new SARS-related coronaviruses, while in the November 2018 grant renewal application EcoHealth only mentioned 52:
We sampled and PCR-screened >16,000 individual bats from 6 families (16 genera) in southern China, finding 9 species positive (5,730 individuals screened) for SARSr-CoVs (Table 1, Fig. 1). We identified 178 novel β-CoVs, of which 172 were novel (52 novel SARSr-CoVs).
It would be interesting to know whether Daszak was just exaggerating or whether EcoHealth has truly identified an additional ~50 novel SARS-like CoVs between November 2018 and December 2019. Judging by the increase in the numbers of sampled bats between the March 2018 DEFUSE proposal and the November 2018 NIH grant renewal application, EcoHealth and WIV have been sampling new bats quite actively:
In particular, they report additionally sampling 1,023 R. pusillus bats, which are the host species of BANAL-103.
It would also be very interesting to know what were the novel SARS-like strains they tested which were able to evade mAbs and vaccines and what they ended up doing with them. Because in the very same December 2019 interview Peter Daszak seems to imply they wanted to use the spike proteins from these strains for SARS vaccines (emphasis added):
[Interviewer]: you’re saying these are diverse coronaviruses and you can’t vaccinate against them, and there are no antivirals, so what do we do?
[Daszak]: …you can manipulate [coronaviruses] in the lab pretty easily — it’s just the spike protein that drives a lot of what happens with zoonotic risk. So you can get the sequence, you can build the protein — and we work with Ralph Baric at UNC to do this — insert it into the backbone of another virus and do some work in the lab. So you can get more predictive when you find a sequence, you’ve got this diversity. Now the logical progression for vaccines is — if you’re going to develop a vaccine for SARS, people are going to use pandemic SARS, but let’s try and insert some of these other related [spikes] and get a better vaccine.
[Interviewer:] I guess also the knowledge of what’s out there, if you see something emerging, it can give it a head start on making a vaccine or therapeutic?
[Daszak]: that’s true
WIV’s interest in SARS1-based immunity escape was quite recent
Against the backdrop of Baric group’s investigation into SARS1-based mAbs and vaccines in 2008, its analysis of SARS1 escape mutants in 2010, and its evaluation of how novel SARS-like CoVs like WIV1 could escape neutralization in 2016, their perennial scientific “coopetitor”, WIV, was actively trying to keep up.
In December 2017, Zhengli Shi co-authored a study titled Cross-neutralization of SARS coronavirus-specific antibodies against bat SARS-like coronaviruses which looked into whether SARS1-based antibodies can protect against WIV1 and SHC014 (essentially, the reference SARS-like bat viruses named in DEFUSE and the NIH grant). It seems that their findings were the inspiration behind the new focus for 2019–2023 on novel SARS-like CoVs whose spikes are 10–25% different from that of SARS1, as those were the strains most likely to evade SARS1-based protection:
In summary, our results have demonstrated that most SARS-CoV RBD-specific antibodies tested in this study could cross-neutralize SL-CoV strain WIV1, but not SHC014. While SARS-CoV and WIV1 have comparable RBD, the RBD of SHC014 is much more variable, as previously noted (Figure S1 in Supporting Information). More specifically, the RBD of SHC014 has a difference of 24 amino acids (aa) compared to that of SARS-CoV, while the RBD of WIV1 only has a difference of 8 aa. This may explain why SHC014 could not be cross-neutralized effectively by most antibodies (mAbs or pAbs) targeting SARS-CoV RBD. The fact that SHC014 retains its ability to infect human cells implies that the available antibodies and vaccines based on SARS-CoV RBD will not protect the next SARS-like disease caused by bat SL-CoV SHC014 strain.
It seems that such strains were sometimes referred to as “highly variable strains” by the grant authors — at least that’s my read based on the quote above (“the RBD of SHC014 is much more variable”) and the description in one of the DEFUSE drafts (emphasis added):
Low abundant mutations, especially in RBD residues that interface with ACE2 receptors, would alter risk assessment calculations as strains identified as low risk, might actually encode high risk, but low abundant variants. To test this hypothesis, we will closely with the modeling core and Dr. Shi’s laboratory to identify highly variable residue changes in the SARSr-CoV S RBD, and use commercial gene blocks to introduce these changes singly and then in combination into the S glycoprotein gene of the parental low risk, high abundant strain parent. We will evaluate the ability of these low abundant chimeric viruses to use human, bat, civet and mouse ACE2 receptors, and more importantly, replicate efficiently in human primary cells.
…
we will synthesize full length rs4237, a highly variable SARSr-CoV that encodes the SHC014 RBD contact interface residues at 442, 487 and 491 but also encodes mutation at 479 (N479S) and has the 432–437 and 458–472 deletions
In light of that, it is quite interesting that based on their January 13, 2020 email exchange — just days after the SARS2 genome was made public — both Ralph Baric and Peter Daszak realized immediately that SARS2 is the “highly variable SARS-like CoV” they set out to hunt for in late 2018:
I wonder if it gave them pause that exactly the virus they decided to hunt for just a year before has now emerged on WIV’s doorstep — and nowhere else — instead of near its natural habitat 1500 km away.
Familiar faces
Coming back to the above 2017 WIV antibody study, it is notable that besides Zhengli Shi its coauthors include Shibo Jiang and Lanying Du. The latter is the widow and close colleague of Yusen Zhou — the SARS and MERS vaccine expert (and reportedly a decorated PLA officer) who filed a patent for a Covid vaccine in February 2020 and then died a few months later under mysterious circumstances.
Shibo Jiang, Lanying Du, and Yusen Zhou have been close colleagues for over a decade, having initially worked on SARS vaccines as far back as at least 2007, when both Lanying Du and Yusen Zhou still held positions with the State Key Laboratory of Pathogen and Biosecurity at the Beijing Institute of Microbiology & Epidemiology. Shibo Jiang’s focus on SARS vaccines and mAbs goes back even farther, as his 2005 and 2006 patents show.
The three researchers have also written extensively on spike-based MERS vaccines from 2013 to 2018, and coauthored joint patents on vaccines against MERS and influenza. By 2019, they have been collaborating for many years under a grant R01AI098775 titled “RBD recombinant protein-based SARS vaccine for biodefense” and have published many papers on SARS and MERS spike-based vaccines:
In 2017, the trio published a paper on a MERS vaccine titled “Recombinant Receptor-Binding Domains of Multiple Middle East Respiratory Syndrome Coronaviruses (MERS-CoVs) Induce Cross-Neutralizing Antibodies against Divergent Human and Camel MERS-CoVs and Antibody Escape Mutants” which implemented the exact strategy that Peter Dazsak talked about in his interview above: the vaccine used multiple RBDs from several MERS-CoVs to generate broader immunity.
In light of the previously mentioned 2017 joint paper with WIV on SARS cross-neutralization, it would be logical to assume that the frequent collaborators were next planning to try the same approach for broad SARS vaccines. In fact, based on this manuscript submitted to biorXiv in May 2020, Lanying Du and Shibo Jiang were indeed working on a SARS vaccine in 2019, under their usual R01AI098775 grant. In that manuscript, the authors mention their interest in the multi-RBD approach for a broad coronavirus vaccine.
Their 2020 paper was a continuation of the work started as far back as 2013, as described in this paper, in which the authors have designed and evaluated several RBD-based SARS vaccine candidates. Intriguingly, in that study led by Shibo Jiang, the authors mention deleting three N-linked glycosylation sites found in the SARS1 RBD:

This is notable because one of those sites — N-40 in the diagram, N357 in the SARS1 spike, mutated to an A in the glycan-ablated mutants RBD193-N3 and RBD219-N3 in the paper above — corresponds to the N370 glycan in SARS2 which mysteriously appears to have been ablated via a T372A mutation not seen in any ancestral SARS2-like strains.
I find this coincidence intriguing because the DEFUSE proposal also suggested ablating some N-linked glycosylation sites in SARS1 and SCH014 to evaluate their role in cross-species transmission and impact on pathogenesis:
SARS S has 23 potential N-linked glycosylation sites N-linked glycosylation sites (NX(S/T); X is anything but proline) and 13 of these have been confirmed using biochemical approaches (e.g., positions: 118, 119, 227, 269, 318, 330, 357, 783, 1056, 1080, 1140, 1155, and 1176). Importantly, several N-linked glycosylation sites regulate SARS particle binding DC-SIGN/L-SIGN, alternative entry receptors for SARS-CoV infections (positions: N109, N118, N119, and especially N227, N699)( PMC1641789, PMC2168787, PMC2168787) and may protect critical sites for antibody neutralization(PMC5515730). Importantly, the emergence of human SARS-CoV from civet and raccoon dog reservoir strains was associated with the evolution of mutations that introduced two N-linked glycosylation sites that promote DC-SIGN/L-SIGN binding (N227, N699), suggesting a role in the expanding human 2003 epidemic (PMC2168787). Interesting[ly], these N-linked glycosylation sites are absent from civet, raccoon dog strains and clade 2 SARSr-CoV, but are present in WIV1, WIV16 and SCH014 as well as human epidemic [SARS-CoV] strains, supporting a potential role in host jumping (Fig C).
To evaluate the role of these mutations in cross species transmission and pathogenesis, we will sequentially introduce clade 2 residues at positions N227 and N699 of SARS-CoV and SCH014 and evaluate virus growth in Vero, Huh7 cells (nonpermissive) expressing ectopically expressed DC-SIGN and HAE cultures, anticipating reduced virus growth efficiency. Using the clade 2 rs4237 molecular clone described above, we will introduce the clade I mutations that introduce N-linked glycosylation sites at positons 227 and N699 and in rs4237 RBD deletion repaired strains, evaluating virus growth efficiency on Vero, HAE or Hela cells ± ectopically expressing DC-Sign (PMC2168787). In vivo, we will evaluate pathogenesis in transgenic ACE2 mice. Experimental outcomes from these studies will be incorporated into risk management models to identify high and low risk SARS-like CoV.
DC-SIGN is a receptor found on the surface of dendritic cells and macrophages, and DC-SIGN binding is relevant in the context of antibody-dependent enhancement of viral entry (e.g. in MERS), which, as I will show later, was a hot topic for WIV and its collaborators in 2019. While the DEFUSE quote above highlights glycans N227 and N699 as important in DC-SIGN binding, the same quote also references another paper (PMC1641789) which instead points to glycans N330 and N357:
We therefore suggest that residues N330 and N357 of the S protein may play important roles for interaction between the S glycoprotein and DC-SIGN.
Thus, it seems that the N357 glycan has been on the radar of the DEFUSE authors and their longtime collaborators, and seeing it ablated in SARS2 is certainly notable.
Of course, the DEFUSE proposal is most notable for mentioning another unique feature of SARS2 — a furin cleavage site, which none of the other known hundreds of SARS-like viruses have. Here is a detailed description of the authors’ interest from one of the earlier drafts of DEFUSE (emphasis added):
After receptor binding, cell surface or endosomal proteases cleave the SARS S glycoprotein to activation fusion mediated entry (PMC4151778). Massive changes in Spike structure occur to mediate membrane fusion and entry (PMC5651768). The absence of S cleavage prevents SARS-coV entry (PMC5457962). A variety of proteases, including TMPRSS2, TMPRSS11a, HAT, trypsin and cathepsin L carry out these processes on the SARS S glycoprotein (PMC3233180, PMC3889862, PMC5479546, PMID:26206723)(Fig C). In some instances, tissue culture adaptations introduce a furin cleavage site, which can direct entry processes as well, usually by cleaving S at positions 757 and 900 in S2 of other coronaviruses, but not SARS (PMID:26206723). For SARS-CoV, a variety key cleavage sites in S have been identified including R667/S668, R678/M679 for trypsin and cathepsin L, respectively, R667 and R792 (and other unidentified sites) for TMPRSS2, and R667 for HAT. Therefore, all SARSr-CoV S gene sequences will be analyzed for the presence of these appropriately conserved proteolytic cleavage sites in S2 and for the presence of potential furin cleavage sites (R-X-[K/R]-R↓) and which can be predicted computationally (PMC3281273). Importantly, SARr-CoV with mismatches in proteolytic cleavage sites can be activated by exogenous trypsin or cathepsin L (Fig D), providing another strategy to recover non-cultivatable viruses. In instances where clear mismatches occur in these S2 proteolytic cleavage sites of SARSr-CoV, we will introduce the appropriate human-specific cleavage sites and evaluate growth potential in Vero and HAE cultures.
The DEFUSE authors’ interest in looking for existing furin cleavage sites in SARS-like viruses and potentially engineering novel ones likely stems from their previous work with MERS: in 2015 Ralph Baric, Shi Zhengli, Shibo Jiang and Lanying Du coauthored a paper showing that engineering a novel FCS in a MERS-like HKU4 virus and ablating a single N-linked glycan enabled it to enter human cells:
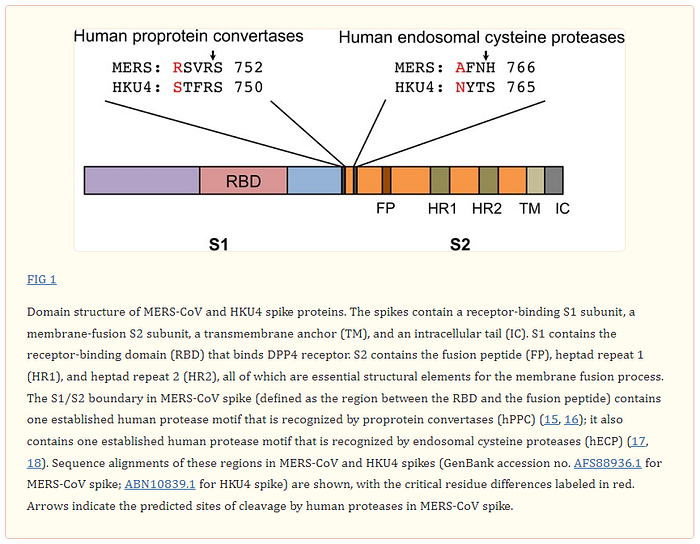
To evaluate the potential genetic changes required for HKU4 to infect human cells, we reengineered HKU4 spike, aiming to build its capacity to mediate viral entry into human cells. To this end, we introduced two single mutations, S746R and N762A, into HKU4 spike. The S746R mutation was expected to restore the hPPC motif in HKU4 spike, whereas the N762A mutation likely disrupted the potential N-linked glycosylation site in the hECP motif in HKU4 spike.
“Restoring the hPPC motif” in the above quote describes restoring the RSVR furin cleavage site found in MERS but absent in HKU4:
It seems that spike protein cleavage, in general, and furin cleavage sites, in particular, were a hot topic in coronavirology in 2018-2019. Other groups were joining in: for example, a Beijing group reported introducing a novel FCS into a chicken CoV in September 2019.
At around the same time, Ralph Baric’s group reported looking into proteolytic cleavage of two new MERS-like CoVs that both had an FCS at the S1/S2 junction. Their findings showed that spike cleavage played a very important role in tissue and species tropism:
Together, these results indicate that proteolytic cleavage of the spike, not receptor binding, is the primary infection barrier for these two group 2c CoVs. Coupled with receptor binding, proteolytic activation offers a new parameter to evaluate the emergence potential of bat CoVs and offers a means to recover previously unrecoverable zoonotic CoV strains.
Surprisingly, despite having coauthored the DEFUSE proposal with Ralph Baric just a year before, Zhengli Shi’s group was not included in the above work. In turn, Ralph Baric’s prior coauthors on the 2015 MERS-HKU4 FCS paper, Zhengli Shi and Lanying Du, did not include his group in their latest 2019 study, but instead were joined by Yusen Zhou to again look into the cleavage of the MERS spike while studying antibody-dependent enhancement of viral entry:
To understand the molecular mechanism of ADE, we investigated whether Mersmab1 triggers any conformational change of MERS-CoV spike. It was shown previously that DPP4 binds to MERS-CoV spike and stabilizes the RBD in the standing-up position, resulting in a weakened spike structure and allowing the S2′ site to become exposed to proteases (51). We repeated this experiment: MERS-CoV pseudoviruses were incubated with DPP4 and then subjected to trypsin cleavage (Fig. 4A). The results showed that during the viral packaging process, virus surface-anchored MERS-CoV spike molecules were cleaved at the S1/S2 site by proprotein convertases; in the absence of DPP4, the spike molecules could not be cleaved further at the S2′ site by trypsin. These data suggest that only the S1/S2 site, and not the S2′ site, was accessible to proteases in the free form of the spike trimer. In the presence of DPP4, a significant amount of MERS-CoV spike molecules were cleaved at the S2′ site by trypsin, indicating that DPP4 binding triggered a conformational change of MERS-CoV spike to expose the S2′ site. Interestingly, we found that Mersmab1 binding also allowed MERS-CoV spike to be cleaved at the S2′ site by trypsin. As a negative control, the SARS-CoV RBD-specific MAb did not trigger MERS-CoV spike to be cleaved at the S2′ site by trypsin. Hence, like DPP4, Mersmab1 triggers a conformational change of MERS-CoV spike to expose the S2′ site for proteolysis.
The authors also looked into how SARS1 entry into immune cells (e.g. macrophages) can be enhanced via an RBD-specific antibody that Shibo Jiang and Yusen Zhou developed previously (emphasis added):
To expand the above-described observations to another coronavirus, we investigated ADE of SARS-CoV entry. We previously identified a SARS-CoV RBD-specific MAb, named 33G4, which binds to the ACE2-binding region of SARS-CoV RBD (49, 50); this MAb was examined for its potential capability to mediate ADE of SARS-CoV entry (Fig. 3E). The result showed that 33G4 mediated SARS-CoV pseudovirus entry into CD32A-expressing cells but blocked SARS-CoV pseudovirus entry into ACE2-expressing cells. Therefore, both the MERS-CoV RBD-specific MAb and the SARS-CoV RBD-specific MAb can mediate the entry of the respective coronavirus into Fc receptor-expressing human cells while blocking the entry of the respective coronavirus into viral-receptor-expressing human cells. For the remainder of this study, we selected the MERS-CoV RBD-specific MAb Mersmab1 for in-depth analysis of ADE.
In the process, the authors ablated the MERS FCS for some of their recombinant spikes:
To stabilize S-e in the prefusion conformation, we followed the procedure from a previous study by introducing mutations to the S1/S2 protease cleavage site (RSVR748–751ASVA) and the S2 region (V1060P and L1061P)
Before that, in 2007 Shibo Jiang, Yusen Zhou, and Lanying Du studied the role of S1/S2 cleavage in SARS1 viral entry (emphasis added):
We further explored if the cleavage of the S protein indeed plays a critical role in SARS-CoV infection. Our results demonstrate that: (1) Factor Xa can effectively cleave the S protein in the pseudovirus SARS-CoV/HIV into S1 and S2 subunits and this cleavage is inhibited by Ben-HCl (Fig. 2B); (2) the S protein can be cleaved into S1 and S2 subunits when the pseudovrus is incubated with its target cells (293T/ACE2) and the level of the cleavage correlates with viral infectivity (Fig. 3); and (3) the target cells express Factor Xa (Fig. 4), a membrane-bound protease [18]. … Thus, the infection of SARS-CoV not only involves the binding of RBD with its receptor ACE2 and the fusion between viral envelope and host cell membrane, but also is associated with the cleavage of the S protein by proteases on the cell membrane, such as Factor Xa.
…
Previous studies have explored several targets for vaccine and antiviral agent developments. For example, vaccine candidates targeting the RBD are able to induce strong neutralizing antibodies and thus provide protection against SARS-CoV infection [19], [20]. Antiviral peptides targeting the HR2 region of the S protein can block the entry of SARS-CoV into target cells [21]. Small molecules targeting SARS-CoV proteases, such as main proteases 3C-like protease (3CLpro) [22], papain-like protease 2 (PLP2) [23] and helicase [24], can inhibit viral replication. In addition to these targets, here we have provided the cleavage of the S protein as another crucial target for the development of vaccines and antiviral agents. Inhibition of the cleavage of the S protein into functional S1 and S2 subunits using agents such as Ben-HCl can effectively block viral entry.
In 2009, Shibo Jiang, Lanying Du, and Yusen Zhou, in the paper titled “The spike protein of SARS-CoV — a target for vaccine and therapeutic development” again talked about SARS1 cleavage, and this time contrasted it with other coronaviruses cleaved by furin:
Unlike the S proteins of coronaviruses cleaved by furin-like proteases, the S protein of SARS-CoV can be cleaved by cathepsin L at position T678 or by trypsin at R667. In contrast to the entrance mechanism of HIV, SARS-CoV can enter cells from an acidic environment of the endosome. Nevertheless, SARS-CoV can also enter the target cell surface, which is mediated by proteases on the cell surface through a non-endosomal-dependent pathway.
In the same paper the authors mention prior work coauthored by Lanying Du on peptides that interfere with S1/S2 cleavage:
Peptides that interfere with the cleavage of S protein. Cleavage of the S protein trimer is an important event in infection, making the potential cleavage site between S1 and S2 domains another target for development of anti-SARS-CoV agents. Synthetic peptides, including P6 (amino acids 598–617) and P8 (amino acids 737–756), both of which are close to the S1–S2 connection and cleavage site, exhibit potent inhibitory activity against the GZ50 strain of SARS-CoV infection in fetal rhesus kidney (FRhK4) cells, and have IC90 values of approximately 100 and 25 μM. This suggests that binding of the peptides to the S protein interferes with the cleavage of S1 and S2, inhibiting the production of functional S1 and S2 subunits and subsequent fusion of the viral envelope and the host cell membrane.
Notably, two years prior to the 2015 MERS-HKU4 FCS study, Shibo Jiang coauthored a paper (for which Lanying Du is listed as the editor) that created a novel RIRR furin cleavage site (albeit not in a coronavirus) via a 12-nt insertion reminiscent of the one that has endowed SARS2 with its RRAR furin cleavage site.
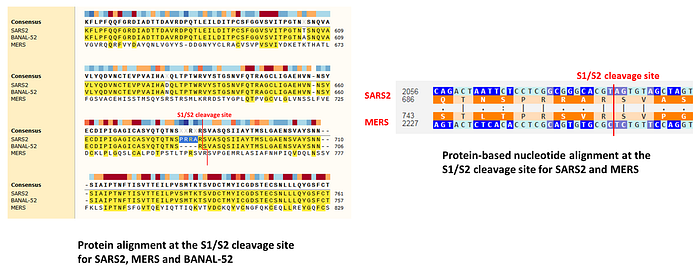
Therefore when SARS2 emerged with its distinctive furin cleavage site —introduced via a very odd 12-nt insertion that also included a leading proline, just as in the MERS FCS — it is very surprising that despite all their prior work, Shibo Jiang, Lanying Du, and Zhengli Shi failed to mention the FCS in their January 2020 paper, despite comparing SARS2 to other SARS-like strains precisely at the S1/S2 cleavage site:
By aligning 2019-nCoV S protein sequence with those of SARS-CoV and several bat-SL-CoVs, we predicted that the cleavage site for generating S1 and S2 subunits is located at R694/S695
What is even more remarkable is that Zhengli Shi’s next major paper in early February 2020, which disclosed RaTG13, also failed to mention the novel FCS — and that paper had 29 authors. It even claimed to provide a protein sequence alignment of the S1 subunit of SARS2’s spike protein relative to RaTG13 and other SARS-like CoVs, but instead of including the full S1 protein, the authors chose to cut their alignment a few residues short of the novel furin cleavage site.
Incredible foresight or a self-fulfilling prophecy?
Looking back at the SARS2 spillover in Wuhan in late 2019, it’s very odd that the virus typically found so far away — in Yunnan or even further south in Laos or Myanmar — decided to spill over right near the Wuhan Institute of Virology. Doubly odd that this happened not long after the institute decided to look for viruses just like it. Triply odd considering that SARS2 has unique features that were never found in SARS-like viruses in nature before or after its outbreak, but were the focus of research interest of WIV and its close collaborators.
So either those researchers had a remarkable gift of foresight and perfect timing in anticipating that a virus like SARS2 is overdue for spillover, which, in a cruel twist of fate, just happened to occur exclusively at their doorstep, or… Or it’s a lab leak.
The latter would be the most parsimonious explanation, as, first of all, WIV was the virus sampling hub for the above researchers, boasting the biggest collection of coronavirus samples in the world: “We sampled and PCR-screened >16,000 individual bats from 6 families (16 genera) in southern China,” said their November 2018 NIH grant renewal application.
Secondly, WIV was also an animal testing hub — they even kept live bats, which at some point Zhengli Shi cared for personally:
Zhang Huajun said: “The research team captured several bats from the wild to prepare as experimental animals, and they need to be fed every day. During the Spring Festival this year, the students went home for the holidays, and Teacher Shi silently took on the task of raising bats.”
WIV also had a flair for doing exotic experiments, such as trying to infect civets by SARS-like viruses, as Zhengli Shi mentioned in her interview:
Q: Has your lab done any animal experiments with SARS-related viruses recently? If so, can you provide any details?
A: We performed in vivo experiments in transgenic (human ACE2 expressing) mice and civets in 2018 and 2019 in the Institute’s biosafety laboratory. The viruses we used were bat SARSr-CoV close to SARS-CoV. Operation of this work was undertaken strictly following the regulations on biosafety management of pathogenic microbes in laboratories in China. The results suggested that bat SARSr-CoV can directly infect civets and can also infect mice with human ACE2 receptors. Yet it showed low pathogenicity in mice and no pathogenicity in civets. These data are being sorted and will be published soon.
Speaking of biosafety regulations, prior to the Covid outbreak, they were pretty lax. In fact, those lax standards at WIV were initially pitched as a selling point for DEFUSE:

Ralph Baric’s comment on this cost-saving measure shows his uneasiness:
[In] the US, these recombinant SARS CoV are studied under BSL3, not BSL2, especially important for those that are able to bind and replicate in primary human cells. In [C]hina, might be growin[g] these virus under [BSL2].
US resea[r]chers will likely freak out.
Heeding Baric’s hint, BLS-2 was changed to BSL-3 in later drafts:

In 2016, however, WIV had no qualms about disclosing in academic papers that work with live SARS-like viruses was done at BSL2 (emphasis added):
Virus and cells
The SL-CoV WIV1 strain (GenBank accession number KF367457) and other viruses were propagated as described previously (2). Sendai virus (SeV) strain Cantell (kindly provided by Hanzhong Wang) was propagated in 10-day-old embryonated chicken eggs at 37°C for 48 h (24). All experiments using live virus was conducted under biosafety level 2 (BSL2) conditions.
They were not alone. Before the pandemic, other labs in China also handled coronaviruses at BSL2. On notable example is a Beijing lab which claims to have isolated the SARS2-like pangolin coronavirus GX/2017 (GX_P2V/pangolin/2017/Guangxi strain) in 2017 and then “routinely” culturing the live virus under BSL2 conditions:
Our reasoning of this isolate having low or no pathogenicity in humans was based on the fact that, back in 2017, no suspected infections were found in those having close contacts with pangolins; and our pangolin coronavirus isolate was routinely cultured in biosafety level 2 facilities.
Finally, as the US instituted a pause on gain-of-function work in 2014, WIV gained another competitive advantage, since it was not subject to US regulations. More and more gain-of-function work kept being done at WIV, culminating in late 2017 with the creation of 8 chimeric SARS-like viruses.
Ancestral habitat of SARS2
The closest strain to SARS2 with a near-identical RBD, BANAL-52, has been found in R. malayanus bats in Laos (incidentally, EcoHealth reported sampling bats in Laos in 2015). Based on extensive bat sampling done after the outbreak, which, in addition to BANAL-52, has yielded multiple SARS2-like strains, the prevailing consensus is that SARS2’s bat progenitor comes from Yunnan or countries bordering it to the south. As Pekar et al. put it:
The closest-inferred ancestors of SARS-CoV-2 likely circulated in Yunnan, China or northern Laos, overlapping with a set of contiguous cave structures extending through these regions.
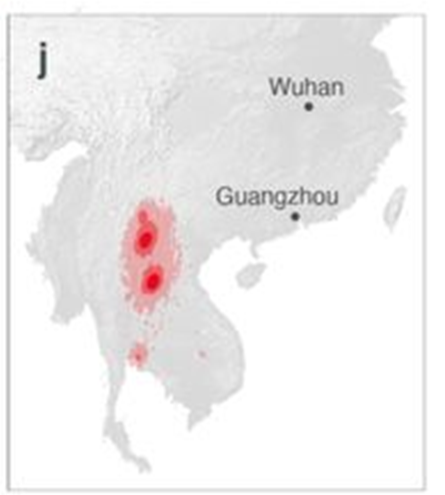
The above authors then try to draw a parallel between the emergence of SARS1 and SARS2:
Our results indicate that both SARS-CoV-1 and SARS-CoV-2 emerged in humans over a thousand kilometers from where their closest-inferred bat virus ancestors likely circulated.
But even according to their analysis, in contrast to SARS2, the bat reservoir of SARS1 was considerably closer to the site of the initial SARS1 spillover in Foshan, just west of Guangzhou (the capital of the Guangdong province):
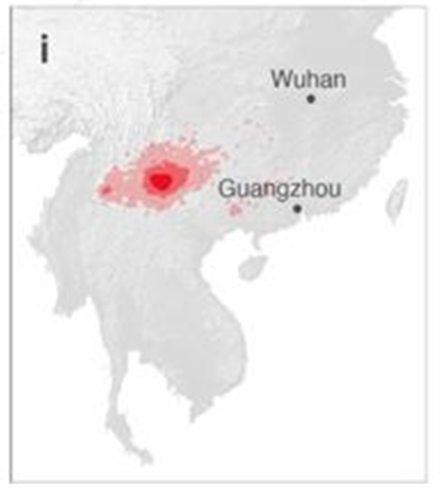
Moreover, the entire Guangdong province lies within a diverse horseshoe bat habitat with many different species carrying SARS-like viruses, while Wuhan is, at best, on the outskirts of that ecosystem:
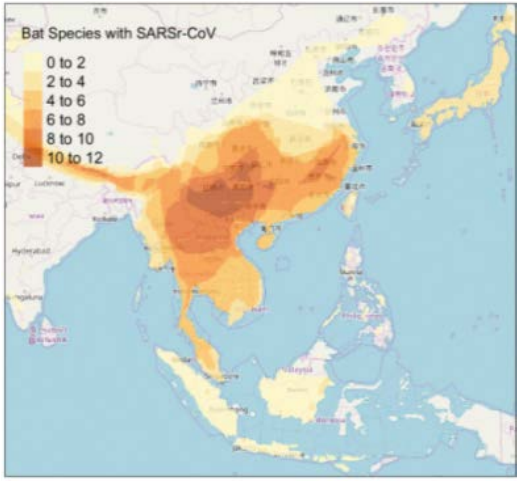
Also, at the time of SARS1 spillover, Guangdong was the center of wildlife trade in China, as well as the capital of exotic culinary practices, as the following 2006 Chinese paper describes (emphasis added):
‘The people take food as their heaven’. ‘Beijing people talk about everything, Shanghai people buy everything and Guangdong people eat everything’. These traditional Chinese sayings illustrate the importance of ‘having good and delicious food’ among the Chinese, especially those from southern China, where the severe acute respiratory syndrome (SARS) epidemic originated. In China, food is considered most nutritive and delicious if it is freshly prepared from live animals, whereas frozen meat is considered inferior. Therefore, wet-markets are located in the vicinity of residential areas in most parts of China, which allows frequent contacts between human and live food animals. In addition to ordinary food animals, many people in southern China, such as Guangdong Province, have the habits of eating a wide range of exotic wild animals, including civets and bats, as this is traditionally believed to improve health and sexual performance.
Wuhan cuisine is much less exotic, and China has cracked down on wildlife trade after the SARS1 outbreak. By the time EcoHealth and WIV were writing their NIH grant renewal application in 2018, they have noted this shift in spillover risk for SARS-like viruses (emphasis added):
Our previous R01 hypothesis was that SARSr-CoV spillover would most likely occur through the trade in bats for food, via the same market chains that [led] to the emergence of SARS (20). To test this, we conducted an exploratory study using standardized one-on-one semi-structured ethnographic interviews and observational data in southern China among 88 people involved in trading wild bats, to assess local social and cultural norms and individual attitudes underlying contact with bats (publication in prep.). Our results suggest that in the 11 years since the emergence of SARS, there have been substantial changes to the wildlife trade: 1) Former wildlife markets are now predominantly selling captive-bred species (poultry, livestock, farmed wildlife); and 2) few bats are now sold through markets. We identified other risk factors for spillover, including people living near to bat roosts, and those visiting bat caves for hunting or recreation.
I can’t help but wonder whether the biggest spillover risk for novel SARS-like viruses in 2019 was now people visiting bat caves for virus hunting.
The SARS2 outbreak is not like that of SARS1
But the most important difference between SARS1 and SARS2 is how they spilled over to humans: the emergence pattern of SARS1 is in stark contrast to that of SARS2. While SARS2 had a single spillover in Wuhan, there were multiple independent spillovers of SARS1 over several months in a number of cities:

In zoonosis, it is much more common for a virus to spill over multiple times, and those spillovers often feature notable genetic differences between strains. Other viruses are also known to have had multiple spillovers. For example, HIV had at least four independent spillovers, while MERS had 50 zoonotic introductions.
During the SARS1 outbreak, scientists quickly found infected intermediate host animals — palm civets and raccoon dogs — and isolated SARS1 strains from them that had many mutations relative to human strains. With SARS2, however, even after 4 years of looking, not a single infected animal has been found.
Zoonosis proponents usually claim that such an animal hasn’t been found for the lack of looking but that is untrue. The search efforts have been considerable. The WHO report on Covid origins goes on for pages and pages detailing such efforts of testing “over 80,000 wildlife, livestock and poultry samples … collected from 31 provinces in China” that were all negative for SARS2:

The Chinese CDC started collecting susceptible animals, including 15 raccoon dogs, around Wuhan as early as January 7, 2020, and in 2022 published their testing results — all negative for SARS2 or its progenitor:
In addition, the now famous Gao et al. study that did detailed environmental sampling of the the Huanan market tested hundreds of samples from many animals sold there but none tested positive for SARS2:
The 457 animal samples mainly collected between January 1 st and March 2 nd , 2020 included 188 individuals belonging to 18 species (with some stray animals sampled until March 30 th ) (Table 2). The sources of the samples include unsold goods kept in refrigerators and freezers in the stalls of HSM, and goods kept in warehouses and refrigerators related to the HSM. Samples from stray animals in the market were also collected, i.e. swab samples from 10 stray cats, 27 cat feces, one dog, one weasel, and 10 rats. All the 457 animal samples tested negative for SARS-CoV-2 nucleic acid, suggesting that the animal infections with SARS-CoV-2 might be rare in the market.
Another study looked at game animals from across China and found 45 new mammalian viruses but no SARS2 progenitor:
Game animals are wildlife species often traded and consumed as exotic food, and are potential reservoirs for SARS-CoV and SARS-CoV-2. We performed a meta-transcriptomic analysis of 1725 game animals, representing 16 species and five mammalian orders, sampled across China. From this we identified 71 mammalian viruses, with 45 described for the first time.
That study also included some pangolin samples:
Previously, a 2020 study focusing on pangolins entering the wildlife trade didn’t find SARS2 either:
Pangolins
Pangolins were an early suspect because the closest case of a pre-outbreak animal carrying a SARS2-like virus — with a near-identical RBD but without an FCS — is a case of infected pangolins reported in 2019. The only caveat is that this case, as well as the presence of SARS-like viruses in those pangolins, were first reported by the Guangdong Institute of Applied Biological Resources (GIABR), a close collaborator of the Wuhan Institute of Virology.
GIABR submitted their paper in September 2019. In it they described sequencing tissue samples collected earlier in 2019 from confiscated pangolins, and reported finding coronavirus genome fragments that matched to a number of SARS-like viruses. Most notably, in the “lung08” sample they reported a 500-nt fragment that aligned to the spike of the HKU3 virus, with a 82.6% identity. HKU3, of course, was listed as one of EcoHealth/WIV’s viruses of interest in the DEFUSE proposal and the November 2018 NIH grant renewal application.
After the outbreak, metagenomic analysis of the sequencing reads from the same “lung08” sample would show that it contained reads matching the SARS2 receptor binding motif (RBM, the part of the RBD that comes into contact with the ACE2 receptor), and pooled reads from lung07, lung08, and lung09 samples would later produce a near-complete genome of the MP789 virus that shares 97% identity with SARS2’s RBD but, more importantly, has a near-identical RBM with SARS2: they share 77 out of 78 residues, including all six residues critical for efficient binding to ACE2.
The senior author of that 2019 GIABR paper on pangolin CoVs, Jinping Chen, collaborated with WIV and EcoHealth before — for example, on a 2014 paper sampling multiple bat species at the Xishuangbanna Tropical Botanical Garden in Yunnan, near the border with Laos, within a few kilometers of where a very notable SARS-like virus, RmYN02, would be found in 2019. So it is quite possible that WIV was aware of GIABR’s findings regarding identifying a number of SARS-like viruses in pangolin samples. In that case WIV would have likely had access to those samples and/or sequencing data in 2019.
According to the declassified US Intelligence (ODNI) report on Covid origins, in 2019, WIV was analyzing at least some pangolin samples (emphasis added):
• Since 2019, some WIV researchers analyzed pangolin samples to better understand disease outbreaks in these animals.
…
• As of January 2019, WIV researchers performed SARS-like coronavirus experiments in BSL-2 laboratories, despite acknowledgements going back to 2017 of these virus’ ability to directly infect humans through their spike protein and early 2019 warnings of the danger of this practice. Separately, the WIV’s plan to conduct analysis of potential epidemic viruses from pangolin samples in fall 2019, suggests the researchers sought to isolate live viruses.
Speaking of the ODNI report, while it seemed to downplay the observations that some WIV researchers “were ill in Fall 2019 with symptoms… consistent with but not diagnostic of COVID-19”, it is still notable that news sources named some members of the Zhengli Shi lab as the ill researchers in question:
The current and former U.S. officials told the Journal the three who fell ill were [Ben] Hu; Yu Ping, a Chinese scientist who wrote a 2019 thesis on SARS-related coronaviruses found in bats; and another scientist named Yan Zhu.
The researchers’ names were noted last week in an article in Public, which publishes on the Substack platform, and were independently confirmed by the [Wall Street] Journal.
In December 2017, Ben Hu — who was a part of the DEFUSE proposal and was the first author of the 2017 WIV paper that reported creating several chimeric SARS-like viruses — gave a detailed interview that echoed themes common with the March 2018 DEFUSE proposal and the November 2018 NIH grant renewal application:
Bioinformatics: After the publication of this research work which lasted for 5 years, what is the next research plan of the group?
Dr. Ben Hu: In addition to the two aspects mentioned above, namely serological investigation for exposed populations and assessment of cross-species transmission of SARS-like coronavirus through animal models, we will not stop the surveillance carried out for bat coronavirus. There is a need to investigate whether unknown strains of SARS-like coronaviruses that pose a potential threat to public health are also present in other bat caves in Yunnan or in other provinces.
Ben Hu previously worked with GIABR on a number of projects. In the same interview he thanked Libiao Zhang of GIABR “who collected a large number of samples for us across the country”:

Intriguingly, in 2019, Libiao Zhang’s team was sequencing bat samples they previously collected in Mengla County in Yunnan to show the presence of Rhinolophus malayanus bats in China, which previously have not been observed there:
The very same bat species would later turn out to be the host of the closest SARS2 relative bat virus found so far, BANAL-52, sampled in Laos, which, along with the MP789/GD-2019 pangolin CoV found in the sequences reported by GIABR, has a near-identical RBD to SARS2. At present, in China, R. malayanus bats are believed to only be found in the south of Yunnan:

As I mentioned previously, EcoHealth and WIV did sample bats in Laos in 2015, about 200 km from where BANAL-52 was found in 2020. In 2021, WIV published a joint paper about this with Libiao Zhang:
While BANAL viruses were found in Laos in 2020 (about 500 km from Mengla County but within the same bat ecosystem), in 2019, another relative of SARS2, called RmYN02, was found in R. malayanus bats in Mengla County just 15 minutes away from the site where Libiao Zhang’s team collected their samples for their 2019 R. malayanus paper:
RmYN02 is notable because it contains a somewhat similar amino acid sequence (PAAR) at the same S1/S2 spot where SARS2 contains a novel FCS (PRRAR). RmYN02’s discoverers even erroneously claimed that, like the PRRA insertion in SARS2, the PAA fragment is also an insertion in RmYN02. However, as more viruses with the same fragment (e.g. BANAL-116 and BANAL-247) were found, we have subsequently shown that the RmYN02 discoverers were incorrect, and PAA in RmYN02 arose via point mutations rather than an insertion, and, judging by the diversity in that fragment between geographically dispersed strains, it arose in their ancestor dozens if not hundreds of years ago.
How did SARS2 get its furin cleavage site?
However, the PAA fragment of RmYN02 is notable because it could have potentially served as an inspiration for a genetic engineer looking to study mutations which could lead to cross-species transmission — in particular, to model what would happen if PAAR mutates into PRRAR to create a novel furin cleavage site that is well-known to expand viral species tropism.
What lends credence to this hypothesis is the fact that the PAA fragment in RmYN02, as well as in BANAL-116 and BANAL-247 strains, is coded by CCT GCA GCG codons, while the PRRA insertion in SARS2 is coded by CCT CGG CGG GCA — i.e. the codons in the SARS2 insertion coding for proline (CCT) and alanine (GCA) are identical to those in RmYN02 and the BANAL strains.
In other words, the PRRA SARS2 insertion looks as if someone took PA from the naturally found PAAR fragment at the S1/S2 spike junction of RmYN02/BANAL strains and inserted two CGG-coded arginines to yield a PRRA insertion, which has, in turn, created a PRRAR furin cleavage site reminiscent of the PRVSR one in MERS.
Some think the above steps could happen naturally, but that is astronomically unlikely. Now, PAAR could theoretically mutate into PRRAR naturally in RmYN02-like strains — although we still haven’t found a single SARS-like bat CoV with an FCS, indicating that there is a strong selective pressure against them in bats — but for the resulting strain to then recombine with a SARS2 progenitor in a way that would only result in the 12-nt PRRA insertion is a statistical impossibility.
In any case, by now, even staunch zoonosis proponents agree that the FCS is extremely unlikely to have arisen in bats, as it is most likely detrimental to enteric tropism, which is favored by these viruses in their natural hosts. However, rather than concede that a genetic engineer might have chosen to engineer the FCS in SARS2, lab leak critics usually respond with some variation of the “but why RRAR?” argument implying that it is less efficient than a canonical RxRR used in some previous FCS genetic engineering efforts.
However, in the past, some researchers actually did choose to engineer non-canonical furin cleavage sites in coronaviruses, for example, RRSR in 2011. Also, in addition to the potential PAAR->PRRAR modeling scenario above, other reasons for RRAR include having observed an RRAR FCS in other sampled coronaviruses, as Zhengli Shi’s team did in 2017 with AcCoV-JC34. Also, WIV has previously identified RRAR as a nuclear localization signal, and coincidentally the first two arginines in that fragment are also coded by CGG CGG.
The potential link to MERS and feline CoV
As to why anyone would engineer a less efficient FCS than RxRR — it turns out, a highly cleavable FCS could actually be detrimental to viral pathogenicity and/or fitness. There is an intriguing Twitter thread about this, which describes the following study of feline coronaviruses that suggests that what turns a benign FECV infection into a lethal FIPV one is the mutation of a highly cleavable RRSRR FCS into something less efficient, like PRRAR:
The study mentions a striking example of two cats living in the same household who were both carrying the benign version of the virus but when one of the cats developed FIP, it turned out that the virus it carried mutated its FCS from RxRR to a less efficient RxxR:

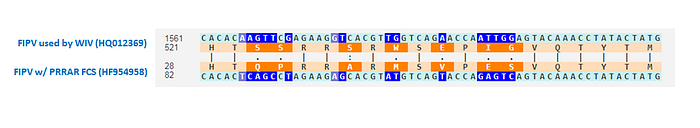
Zhengli Shi must have been familiar with this aspect of feline CoVs. In 2015, she coauthored a joint paper between WIV and Wuhan University that tested a pan-coronavirus inhibitor against several CoVs, including SARS1, MERS, and FIPV. The FIPV strain they used had the same FCS mutation as above, which turned a canonical RxRR FCS into a less efficient RRSR :
The author of the Twitter thread who pointed out the paper on FCS mutations turning FECV into FIPV did so in connection with the 2019 WIV paper on MERS antibody-dependent enhancement (ADE) of viral entry that I alluded to before. The connection is that feline CoVs are also known to exhibit ADE, and the 2019 WIV paper references three papers on feline CoV ADE (emphasis added):
ADE has been observed for coronaviruses. Several studies have shown that sera induced by SARS-CoV spike enhance viral entry into Fc receptor-expressing cells (42,–44). Further, one study demonstrated that unlike receptor-dependent viral entry, serum-dependent SARS-CoV entry does not go through the endosome pathway (44). Additionally, it has long been known that immunization of cats with feline coronavirus spike leads to worsened future infection due to the induction of infection-enhancing antibodies (45,–47). However, detailed molecular mechanisms for ADE of coronavirus entry are still unknown. We previously discovered a monoclonal antibody (MAb) (named Mersmab1) which has strong binding affinity for MERS-CoV RBD and efficiently neutralizes MERS-CoV entry by outcompeting DPP4 (48); this discovery allowed us to comparatively study the molecular mechanisms for antibody-dependent and receptor-dependent viral entries.
As you might recall, in that 2019 WIV paper, Zhengli Shi, Lanying Du, and Yusen Zhou looked into how antibody binding could enhance MERS viral entry into immune cells via modulating spike cleavage. In light of that, parallels between the furin cleavage sites of MERS (PRSVR), SARS2 (PRRAR) and some of the above FIPV CoVs (PRRAR) are quite interesting.
The desire to equip a SARS2 progenitor with a MERS-like FCS can also explain why a genetic engineer might have chosen to do so via an insertion rather than mutation of existing nucleotides: the MERS spike protein is four residues longer at the S1/S2 junction than SARS-like viruses, and an insertion like PRRA removes this imbalance, and potentially helps fully align the 3D structure of the two proteins at this junction:
Recall that as part of their 2019 MERS ADE study, WIV and colleagues were already looking at antibody-dependent enhancement of SARS1 and claimed that a mAb they previously developed can help SARS1 get into immune cells:
To expand the above-described observations to another coronavirus, we investigated ADE of SARS-CoV entry. We previously identified a SARS-CoV RBD-specific MAb, named 33G4, which binds to the ACE2-binding region of SARS-CoV RBD (49, 50); this MAb was examined for its potential capability to mediate ADE of SARS-CoV entry (Fig. 3E). The result showed that 33G4 mediated SARS-CoV pseudovirus entry into CD32A-expressing cells but blocked SARS-CoV pseudovirus entry into ACE2-expressing cells. Therefore, both the MERS-CoV RBD-specific MAb and the SARS-CoV RBD-specific MAb can mediate the entry of the respective coronavirus into Fc receptor-expressing human cells while blocking the entry of the respective coronavirus into viral-receptor-expressing human cells.
A researcher looking to expand that research to novel SARS-like CoVs could then do something similar to a SARS2 progenitor, which could explain the unique SARS2 mutations like R346 and A372. The first one is reminiscent of a SARS1 escape mutation from the S109.8 antibody, while the second removes an N-linked glycan previously implicated in DC-SIGN binding (a receptor on immune cells involved in ADE).
Coming back to the FCS itself, a genetic engineer with a recently stated interest — as per the March 2018 DEFUSE proposal — to look for existing furin cleavage sites in novel SARS-like viruses and create new ones, could have had a number of reasons to create an FCS like the one seen in SARS2. Moreover, such an engineer might have then had several different FCS candidates of varying furin cleavage efficiency, and evaluated how such efficiency affected tropism, pathogenicity, transmissibility and ADE.
Mengla County
How likely was WIV to have come across a BANAL-like and/or RmYN02-like strain prior to the SARS2 outbreak? Quite likely. Not only did WIV sample in Laos, but they have also done extensive sampling in Mengla County, as evidenced by a 2017 paper, as well as a table below from a 2018 paper coauthored by Ben Hu and Zhengli Shi:
In 2019, Zhengli Shi even reported a novel virus they discovered in Mengla County which they named “Mengla Virus”. In 2019, they fully sequenced that virus from samples collected in December 2015. Ben Hu is not on that paper but Yan Zhu (one of the 3 WIV staffers named as sick with Covid-like symptoms in Fall 2019) is, as is Libiao Zhang.
Also, in January 2019, Shi’s team and Libiao Zhang returned to Mengla County to sample more bats in 2 caves and track them via GPS. They published their results in 2022, showing cross-border incursions of Mengla bats into neighboring Laos and Myanmar:

Overall, the above observations paint a compelling picture of extensive sampling by WIV and its collaborators of the bat ecosystem widely believed to house the bat progenitor of SARS2:

Curiously, before carrying out the GPS bat tracking research of Mengla bats in 2019, WIV pitched putting GPS trackers on bats in the DEFUSE proposal:
ICARUS satellite transmitters (1g) wil be attached to 12 Rhinolophus spp. bats from each study roost (36 bats total) to determine nightly foraging dispersal patterns
Which goes to show that just because DEFUSE wasn’t funded doesn’t mean WIV couldn’t do the proposed research on their own. After all, WIV had ample funding from Chinese sources. For example, in 2018, Ben Hu was awarded a three-year grant starting from January 2019 titled “Pathogenicity of Two New Bat SARS-Related Coronaviruses to Transgenic Mice Expressing Human ACE2 Receptor.” His boss, Zhengli Shi, also had a number of local grants, including a large 8.5mm RMB (~$1.2mm) grant to study “Genetic evolution and transmission mechanism of important bat-borne viruses,” among others:
But what about the Huanan Seafood Market?
The entire case for the natural origin of Covid — which posits that the proximity of the SARS2 outbreak to WIV is a mere coincidence — is built on the claim that SARS2 spilled over to humans at the Huanan Seafood Market via some unknown intermediate animal. “Why are the early Wuhan Covid cases centered around the market if the virus didn’t emerge there?” is the essence of the zoonotic hypothesis.
However, a wildlife market is the perfect location for a novel virus to have gotten noticed in China, even if that virus came to the market from a nearby lab. First, wildlife markets are great superspreading locations for SARS2, as we learned during the course of the pandemic: in several instances such markets sparked Covid outbreaks in places previously Covid-free: in Thailand, Singapore, and twice in China. The Huanan market was a very busy place — 10,000 daily visitors spiking up to 100,000 on the weekends — near a key transportation hub in Wuhan.
But more importantly, after the SARS1 outbreak, contact with wildlife was a big red flag in the Chinese surveillance system for novel “unexplained pneumonias”. Annual pneumonia outbreaks are common, and without additional criteria it would be impractical to treat every cluster of pneumonia cases as a signal of a novel coronavirus. So if a SARS-like virus leaks from a lab during a flu season, it could well remain undetected before arriving in a wildlife market and infecting several elderly people severely enough to end up in a hospital, which would then report this cluster of suspicious cases to the centralized surveillance system.
After that, of course, the market would become the prime suspect, which is exactly what happened. So much so that initially a market link was a part of the diagnostic and reporting criteria for Covid. Unfortunately, even after the initial fog of war cleared, a proper retrospective search for the earliest patients was never carried out. If you are interested in diving deeper into the history of early cases, as well as learning about a potential connection of some market patients with a Mahjong room at the market, I recommend this excellent presentation by Gilles Demaneuf. As things stand right now, the current subset of known early cases exhibits — quite understandably — signs of a pro-market ascertainment bias.
The market was dominated by a later lineage of the virus
The biggest problem for the market origin hypothesis is that all 16 of the earliest Covid patients linked to the market were infected by lineage B of the virus — a phylogenetically later version than ancestral lineage A also circulating in Wuhan at the time. This is problematic for the zoonotic hypothesis because in a typical spillover event one would expect the earliest strain of the virus dominating the site of its spillover.
Additionally, nearly all environmental samples from the market that were sequenced extensively enough to determine their lineage were identified as lineage B. The only lineage A sample later detected in the market did not come directly from an environmental swab but rather from a cultured laboratory experiment attempting to grow the virus from that swab. Despite the original environmental sample having a very high Ct PCR value and containing only 22 SARS2 genomic sequencing reads, indicating a low viral load in that sample, a virus was successfully cultured from it. Surprisingly, the cultured virus had additional mutations not present in early genomes but observed later in the pandemic, suggesting possible laboratory contamination rather than genuine market origin.
In any case, the overwhelming dominance of lineage B in both early human cases and environmental samples from the market strongly implies that the market was not where SARS2 initially jumped to humans. Instead, the market likely amplified a lineage B strain sometime after lineage A was already in circulation in Wuhan. This scenario is further strengthened by the observation that lineage B potentially had an edge over lineage A in terms of transmissibility, as experimentally lineage A samples replicated to lower titers than lineage B ones.
The double spillover hypothesis
To explain the dominance of lineage B in the market that undermines the zoonotic spillover hypothesis, one pro-zoonosis research group proposed that the two lineages actually represent two separate spillover events from wildlife. In that scenario, an animal with a phylogenetically later strain, lineage B, was brought to the market first, started the Covid outbreak, and only then, a second animal carrying an earlier, ancestral strain of the virus arrived and started infecting humans with lineage A. Of course, even that scenario doesn’t explain why all 16 early patients from the market had lineage B.
Before detailing how the Pekar et al. two-spillover hypothesis turned out to be based on incorrect assumptions and erroneous modeling code, I should say that even if the two SARS2 lineages truly stemmed from two spillovers, that does not preclude a lab leak, as it is entirely possible for a virus to leak from a lab more than once. In fact, SARS1 escaped from the same lab twice in a span of two weeks in 2004, if not thrice.
The Pekar et al. hypothesis was predicated on two things: first, the authors disregarded all known intermediate genomes between A and B under the explanation that they all stemmed from sequencing errors, rather than were true descendants of lineage A. Then the authors developed a Bayesian model to simulate viral evolution, providing probability estimates for different ancestral scenarios.
In their initial paper, this model indicated a fairly strong Bayes factor in favor of two separate spillovers. But thanks to some remarkable citizen science, it turned out the Pekar et al. code had a number of bugs. Once those bugs were fixed, the Bayes factor supporting two spillovers materially decreased:
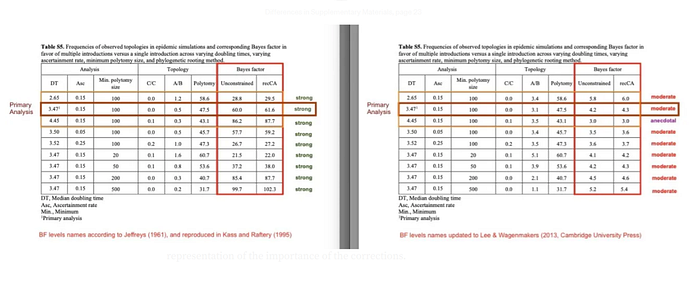
That alone severely undermined the double spillover hypothesis. However, what provided dispositive evidence for rejecting it, was confirming the existence of true intermediate early genomes between lineages A and B (called B0 in the quote and figure below):
Notably, two B0 genomes recovered here and seven genomes recovered from Wuhan, Singapore, and the UAE were at the basal position of lineage B and its descendants (abbreviated as Lineage Bs).
…
Lineage B0 viruses (haplotype ‘T/T’) were reported in Wuhan, Singapore, and the UAE (Supplementary Table S3) and occupied the intermediate position of lineage A and lineage B. Herein, lineage B0 genomes were recovered from two patients, who were infected at different regions (Henan and Shanghai) and hospitalized on 4 and 8 February, respectively. Interestingly, our genomes share 100 per cent similarity with four genomes sampled from Wuhan and Singapore. Overall, these genomes have no difference from the reference genome of lineage A or B with the exception of the nucleotide at the site 8,782 or 28,144, but it should be noted that there was an indeterminate C/A nucleotide assignment (1,125 counts of ‘C’ and 2,008 counts of ‘A’) at the position 27,230 of SH-P37–2-Shanghai genome. Finally, either lineage A0 or B0 genomes were recovered and confirmed by both RT-PCR and transcriptomic protocol (SRR25229357, SRR25229358, SRR25229360, and SRR25229361). Hence, these data indicate the true existence and evolution of lineages A0 and B0 viruses (Supplementary Table S8) in human populations.
Furthermore, even lineage A might not have been the original lineage that spilled over to humans. Some of the earliest genomes were found to be even closer to the bat ancestor than lineage A — for example, A0 (with a C29095T mutation) in the above paper. The same mutation was observed in one of the early Wuhan patients, in a sequencing dataset subsequently removed from NCBI but recovered by Jesse Bloom. That dataset also contained sequences from several more patients that likely had lineage A.
In conclusion, there is no reason to believe there were two separate spillovers. The scenario most consistent with known observations is that lineage A evolved into lineage B already in humans. This strongly implies that the Huanan market was exposed to a subset of genomes that emerged after the initial spillover, and therefore was not the initial spillover site but rather a location where an already circulating lineage B was amplified.
Steelmanning zoonosis vs. lab leak
Let us now digest the observations described above and produce two strongest scenarios for each origin hypothesis. Then you, dear reader, can decide for yourself which one sounds more plausible.
For that I will defer to Will Van Treuren who served as a judge in the Rootclaim Covid origins debate. The two judges in that debate ruled against the lab leak hypothesis, so I am unbiased here in endorsing the scenarios Will has constructed — even if I believe that he picked the wrong one (possibly because at the time of the debate we didn’t yet have the DEFUSE drafts or conclusive evidence of intermediate genomes ruling out double spillover). Here are the two scenarios, extracted from his debate decision:

My view
While I appreciate that through the prism of their knowledge, experience or biases, different people might weigh known evidence differently in favor of one or the other hypothesis, I think a lab leak is the more parsimonious explanation. I find zoonosis much less likely because I just can’t get over the series of incredible coincidences it requires to explain the location and the timing of the outbreak.
Not only is there no convincing evidence of zoonosis four years after the outbreak, even the best hypothetical scenario outlined by Will of how an intermediate host would get to Wuhan, and only Wuhan, via wildlife trade is implausible, in my view. Exclusivity of Wuhan aside, years before getting to Wuhan, that animal host needed to have picked up an enteric bat virus, only transmissible via an oral-fecal route, sustained it in its population long enough to develop an FCS for that virus to become respiratory, and thus highly transmissible, but then waited until that animal traveled to Wuhan to only infect people once it got there but not along the way — and have that exclusive journey happen twice in a row in a span of a week or two, as postulated by the double spillover scenario that is required to reconcile the contradiction of lineage B dominating the market.
That scenario requires hundreds if not thousands of animals sustaining a reservoir of the virus for years, so we would expect to have discovered numerous infected animals by now, much like during the SARS1 outbreak where several strains were identified in palm civets and raccoon dogs with sequencing technology from 20 years ago. The failure to detect any such infected animals today, despite vastly improved sequencing capabilities, severely undermines zoonosis.
Conversely, on the lab leak side, there is a whole range of plausible scenarios. A lab leak doesn’t even require a single lab member knowing about it — the virus could have leaked during virus culturing experiments. If someone in Wuhan was culturing pangolin samples from the same batch as the one sequenced by GIABR in October 2019, later found to contain the MP789 virus, or even Mengla samples in search of the Mengla virus, a SARS2 progenitor contaminating those samples could have been — unknowingly — successfully cultured, and then leaked due to the culturing being done under BSL2.
Alternatively, as Will’s scenario describes, a lab leak could have occurred from the research of a single grad student pursuing the directions outlined in DEFUSE and the EcoHealth/WIV NIH grant, while everyone else including his/her superiors could plausibly plead ignorance. Finally, the research resulting in a lab leak could have stemmed from the research into antibody-dependent enhancement in connection with broad RBD-based vaccines for SARS-like and MERS-like viruses, as well as other antibody-based and drug-based pan-coronavirus therapies.
It is that type of research that WIV was actively pursuing in late 2019, with several collaborators in China and the US, including EcoHealth. And it is that type of research for which SARS2 fits the profile of a novel SARS-like virus that WIV has been on the hunt for since at least 2018.
So when a virus fitting that profile spills over exclusively in Wuhan and contains unique features that WIV talked about in research proposals shortly before, and four years later we have not found its animal progenitor or a single infected animal, the most plausible explanation to me is that it was a lab leak.
Epilogue
Four years later — to the day — since I published my first Medium article exploring Covid origins, the case for zoonosis only got weaker, while the case for lab leak got stronger.
Several factors have weakened the zoonotic hypothesis. Probably the biggest factor is not finding any infected intermediate animals. On the academic side, the paper hailed as a “lab leak killer” in 2020 is now facing calls for retraction (there is even a petition) after internal Slack messages have shown that its authors held very different views in private. In intermediate years, claims of “dispositive evidence” of the Huanan market being the site of spillover came and went, as did the double spillover hypothesis.
Strengthening the lab leak side, the DEFUSE proposal was revealed in mid-2021, and its early drafts were later released via FOIA. In addition, the EcoHealth/WIV NIH grant renewal application provided additional color as to the type of research WIV was doing in 2019, as well as the kind of SARS-like viruses they were searching for.
So today, while we do not yet have certainty in any particular Covid origin scenario, a lab leak origin looks highly likely. Hopefully, within another four years, this mystery will be solved completely. Until then, we can keep debating about raccoon dogs, pangolins, and stuffy Mahjong rooms at the Huanan Seafood Market.
