Why Ralph Baric’s Testimony Strengthens the Case for a COVID-19 Lab Origin

Highlights:
- Ralph Baric confirmed that DEFUSE proposed inserting novel furin cleavage sites into live viruses, inspired by feline coronaviruses
- SARS-CoV-2’s furin cleavage sites is identical to the one found in several lethal feline coronavirus strains
- Ralph Baric strongly believes WIV had unpublished viruses and viral reverse genetics systems
- Most plausible Covid origin is the result of research on identifying new SARS-like viruses and developing broad vaccines against them
Quick Summary:
Ralph Baric’s January 2024 testimony provided crucial insights into the DEFUSE grant proposal, a collaborative project involving EcoHealth and the Wuhan Institute of Virology (WIV), and the research interests of WIV prior to the Covid outbreak. The pre-Covid landscape of coronavirology research saw intense US-China collaboration aimed at creating broad-spectrum vaccines for SARS- and MERS-like viruses. Key players like Baric, along with researchers Lanying Du, Yusen Zhou, Fang Li, and Shibo Jiang, focused on overcoming challenges such as antibody-dependent enhancement (ADE). By 2018–2019, as collaboration between Baric and his counterparts shifted, WIV became central in SARS-like virus research.
Drawing on Baric’s testimony, I argue that the emergence of SARS-CoV-2 in Wuhan wasn’t coincidental but a likely result of research at WIV. In particular, I outline how WIV researchers, after a post-2018 shift in focus, concentrated on identifying SARS-like viruses with spike proteins 10–25% different from SARS1, capable of evading SARS1-based antibodies, and potentially causing ADE. They were plausibly inspired by Baric’s ideas on furin cleavage sites and his work with Fang Li on SARS and MERS spike cleavage, leading them to engineer furin cleavage sites into novel SARS-like strains. Baric’s testimony suggests that feline coronaviruses inspired this suggestion, and the identical furin cleavage sites in lethal feline coronavirus strains and SARS-CoV-2 strongly indicate that the WIV could have inserted such a site into a SARS-CoV-2 precursor to extend their research from MERS to novel SARS-like coronaviruses. This suggests the emergence of SARS-CoV-2 was not a coincidence but a potential result of this focused research.
In his recently published testimony from January 2024, Ralph Baric, the preeminent coronavirus expert, dispelled several talking points that lab leak critics have used repeatedly. He confirmed that the DEFUSE grant proposal — coauthored by him, EcoHealth, and the Wuhan Institute of Virology — was indeed suggesting to insert novel furin cleavage sites into SARS-like viruses, and do so in live full-length viruses rather than just pseudoviruses or chimeras. Importantly, he also said that the WIV not only had unpublished viruses but almost certainly had unpublished molecular clones (i.e. viral reverse genetics systems).
Moreover, Dr. Baric strengthened a plausible explanation for why a genetic engineer might have chosen to create a furin cleavage site like that in SARS2 — to mimic one often seen in pathogenic strains of a feline coronavirus. As I will show in this article, this explanation fits well with one plausible lab leak scenario: that SARS2 leaked during work on novel SARS vaccines conducted by Yusen Zhou — a SARS and MERS vaccine expert (and reportedly a decorated PLA officer) who filed a patent for a Covid vaccine in February 2020 and died a few months later under mysterious circumstances.
Yusen Zhou was married to Lanying Du, a frequent collaborator of Shibo Jiang, Zhengli Shi, and Fang Li. As far back as 2016, the WIV website called Fang Li a visiting researcher at WIV when showcasing their paper on MERS vaccines where Lanying Du is the first author, while Shibo Jiang, Yusen Zhou, and Fang Li are all senior and corresponding authors.

Feline coronaviruses, their mutating furin cleavage sites, and how such mutations enable viruses to infect immune cells and cause antibody-dependent enhancement of viral entry (ADE) is the thread that connects the research described in DEFUSE and EcoHealth grants to the research that Yusen Zhou, Lanying Du, Shibo Jiang, Zhengli Shi, and Fang Li were doing independently — without Ralph Baric or EcoHealth — in the context of MERS and SARS in late 2019.
But before I dive into their research to outline how a genetic engineer might have arrived at the idea to insert a furin cleavage site from a feline coronavirus into a SARS2 progenitor and potentially study its impact on ADE, let me first turn to Dr. Baric to set the stage. I believe his next few quotes are very important to understanding the key coronavirology research areas in the years leading up to the Covid outbreak:
1. Developing broadly active coronavirus vaccines and therapeutics
Dr. Baric: “One of the things that drove the 2015 paper was that SARS coronavirus emerged in 2003. It was controlled by public health intervention strategies because it didn’t transmit until you got clinical disease. People thought it was a fluke, one-off, it’s not going to happen again. Then MERS coronavirus emerged in 2012, again; highly pathogenic, 35 percent mortality rate, but it didn’t transmit very well. So that data made us ask the fundamental question: What is the risk level that exists in nature? This paper, in essence, said the risk in nature — that risk existed in nature. And then the mouse models were then used to develop countermeasures. So almost immediately in parallel with this paper, we started working with Gilead Scientific to evaluate nucleoside inhibitors that might work against the coronavirus family. After testing a bunch of things, we eventually got down to remdesivir, demonstrating that it worked against the MERS coronavirus and the SARS coronavirus. That led to a companion paper that included these viruses in 2017 that said these are broad spectrum antivirals that work in robust animal models of disease. And the preclinical data was now available to move into the clinical trials. So that’s why animal models are so important. Ultimately, remdesivir, molnupiravir, the Moderna vaccine, I don’t know if we ever did the Janssen vaccine. But several therapeutic antibodies had all made it through the FDA and into the clinic, went through our lab, and many of them touched these viruses that were developed in the 2015 paper. These same viruses are being used for universal vaccine design for all sarbecoviruses and all betacoronaviruses [here and subsequently all emphasis is added by me — Y.D.]. So if you want to really protect the public, you have to have the appropriate virologic reagents that challenge the effectiveness of either your drug or your antibody or your vaccine and prove performance. So ultimately, the goal of what resulted from this paper was the idea that we had to develop drugs, we had to develop immunotherapeutics that were broadly active. And we had to develop vaccines that were broadly active. And that paper, including the viruses, the human viruses that occurred, were included in studies that were used with the Moderna vaccine as well. So, again, animal model development is key to this. It’s, again, very, very easy to make drugs that work against something that barely replicates, but then when they get into the humans, they fail. So that’s the basis for it. That’s probably a little longwinded. I apologize. Anyway, that’s the thought process.”
2. Broad coronavirus vaccines and “vaccine-associated deleterious outcomes” (ADE — antibody-dependent enhancement of viral entry)
Q: “I appreciate that science lesson. I’m going to change topics a bit. We have heard from multiple witnesses that the creation of a vaccine for COVID-19 happened almost miraculously fast, and they credit this speed to the fact that coronavirus research and mRNA research had been going on for years prior to the COVID-19 pandemic. You were a part of this process, both with ongoing research and active involvement in the COVID-19 vaccine testing, correct?”
A: “That’s correct.”
Q: “In terms of the development and testing of a COVID-19 vaccine, in 2020, your involvement was running safety and efficacy trials for Moderna’s vaccine using your lab’s chimeric coronavirus strains, human respiratory cell cultures, and lab mice. Is that accurate?”
A: “For the COVID-19 vaccine, I don’t think we tried any — we used any chimeras. The only thing we really used was the mouse-adapted SARS2 coronavirus, the MA10, which was called MA10 in this case. It was ten passages in mice that produced a lethal infection. But I can tell you that our involvement with mRNA technology started in 2016, early 2017, in collaboration with Barney Graham and Kizzmekia Corbett at the NIH VRC, where they had just worked. Well, Jason McLellan and Barney had really worked out the technology to freeze the coronavirus spike glycoprotein in what was called the prefusion state, which had all the big, juicy neutralization epitopes in the right context. So they wanted to evaluate mRNA vaccine performance, and so they contacted us and we worked with them on mRNA vaccines for MERS coronavirus mostly, but also SARS coronavirus in 2003, and were actually writing the paper in December 2019 when COVID hit. And so we stopped writing the paper. When they received the sequence, they ordered the constructs. I was told that I had to have a mouse model available by the end of April, so my job was to make a robust mouse model in sufficient time to test that vaccine in April and May, so that the final reports could be compiled, including some studies that were designed to look for what are called variant phenotype vaccine associated — oh, crap, I forget the name. Do you have to type everything that I say? Great.”
Q: “We’re all allowed to have those moments.”
A: “I’m having a moment. But they’re probably going to become more frequent over the next hour, I have to admit. But it’s vaccine associated deleterious outcome. In this case, there’s something, either the vaccine enhances the availability of the virus to grow or it causes some kind of pathology. And it needed to be tested for that, because, earlier, it had been shown with earlier vaccines with the SARS strain that you’ve got those phenotypes. My job was to make the mouse model and design those experiments and have them all done by April.”
Q: “And we’ve heard from multiple people that this was all on a timeline that was way faster than any other vaccine.”
A: “It was very stressful.”
Q: “I’m sure.”
A: “It was very stressful.”
Q: “You mentioned that you had been working on this, on vaccines, prior to 2016. I know, reading articles and research that you’ve done, it seems like you’ve been working on a pan-coronavirus vaccine for many years, and that’s been one of your research focuses; is that right?”
A: “Well, again, the discovery work we did said that there was a zoonotic virus. There are animal viruses out there that are high risk. You don’t know which one will evolve. So the only kind of countermeasure you can make is broad spectrum. It either has to be a broad spectrum drug, or you have to have a vaccine that provides like an umbrella of breadth to many strains. And so what you try to do with your discovery work is to find the strains that are the most different, and then some in the middle. So then you can say, well, it works on the bookends, it works in the middle, I hope it works against the new thing, right?”
Q: “Sure.”
A: “That’s the only way to do it.”
Q: “You mentioned a little bit throughout today some therapeutics that you were testing before and other research that was sort of useful for the pandemic. Can you elaborate on what pieces or findings from research prior to the pandemic were useful in determining and finding vaccines and therapeutics once the pandemic was widespread?”
A: “Well, certainly having isolates and robust mouse models of human disease, using the human strain of MERS and the SARS strain that caused human disease were really important. But that captured this much of the variation, like a paper-thin sliver of the variation that exists in the family. So you need to have natural, other zoonotic isolates with robust mouse models, so you’ll be able to really evaluate the performance of the vaccine when it’s not a perfect match, because when the vaccine’s not a perfect match is when all these adverse reactions can occur, or you have this because you have a breakthrough. So we did discovery work. That discovery work is important because it gave us breadth both with MERS and with SARS. In addition, at the same time, we were part of a grant that was funded to try to develop drugs against coronaviruses, with Mark Denison at Vanderbilt and Gilead were collaborators. And so Gilead was gracious enough to provide a fairly robust panel of nucleoside inhibitors that we screened working down to remdesivir, that we then moved from — the classic approach was, you know, cells, continuous cells in culture, to primary human cells, to the animal models, and demonstrated that it not only worked against SARS and MERS but it worked against all these other bat coronaviruses, other human coronaviruses, other animal coronaviruses, 12 different viruses. So we knew it had broad spectrum. So now the hypothesis is, you have a broad spectrum drug. Any new virus comes along, you immediately test the hypothesis and evaluate remdesivir, molnupiravir, Paxlovid, therapeutic antibodies, vaccines, to see if they provide breadth. And simultaneously, you use that information in a reiterative fashion now to develop broader-based vaccine platforms. So one of the innovations that we did was to take spike glycoproteins across the phylogenetic tree, blend them together as a chimera, delivered on mRNA vaccine that would provide neutralizing breadth against a greater percentage of the strains.”
Q: “So would it be accurate to say that research on a pathogen that’s not yet infecting people gives scientists a basis to make their hypotheses for how a pathogen that is infecting people may react to therapeutics or a vaccine?”
A: “It’s more than that, It’s absolutely essential. You have no idea of the breadth of performance of your product if you don’t have natural isolates available in the virus family. So, for example, calls to shut down discovery work in the natural world will basically mean that the U.S. is at greater risk for future emerging diseases because we don’t know what’s there, and we can’t test products against it.”
3. Looking for novel SARS-like viruses whose spike are 20–25% different from SARS1
Q: “And then on the other aspect of looking — and this may relate to sort of the search for a broad spectrum coronavirus vaccine. What was the rationale between looking for a SARS-related coronavirus that sort of a 10 to 20 percent divergent in the spike from SARS1?”
Dr. Baric: “Sure. So SARS 2003 is the bookend, right? You know how much variation. WIV1 and SHCO14 have about 8 to 12 percent variation in the spike or the RBD. The clade 2 strains like HKU3 have 30 to 35 percent variation in the spike, they’ve got deletions in the RBD, they can’t use human ACE2 receptors. If you take those two numbers, subtract 10 or 12 from 35, divided by 2, added to 12, you get a number between 20 and 25. And that was our prediction, that there would be strains with that much variation that could still use human ACE2 receptors. It turns out SARS2 had 22 percent variation, so we were within the range, but we were really not completely right. In MERS, there are strains with 35 percent variation in the RBD that could still use the human. So in reality, it’s probably much greater than 20, 25 percent.”
Q: “Really?”
A: “That was our estimate. And the reason we’re interested in that, the strains with the most variation become important for developing countermeasures in vaccines. So if you have a strain that’s really different than therapeutic antibodies, you can look for broadly neutralizing antibodies. They may not work. Your vaccine, if you have an animal model, you can ask, does it cover this much variation? And if it doesn’t, it gives you the starting material to develop a second generation vaccine that can capture it. So again, that variation — I have no interest in simply resurrecting every single coronavirus.”
Q: “Sure,”
A: “I’m interested in the bookends and a couple intermediate ones because that’s what’s best for countermeasure development.”
Key research areas in coronavirology prior to the Covid outbreak
I hope you read the above quotes fully, because they shed light on what Ralph Baric and, by extension, his collabo-competitors were interested in in the years preceding the outbreak: working on broad vaccines and therapeutics that could protect against many coronaviruses, especially SARS- and MERS-like ones. To get the right breadth of SARS-like CoVs, by 2018, Baric, along with WIV and EcoHealth, were now looking for novel strains that were 20–25% different from SARS1 in their spike protein (SARS2 is 24% different).
However, one challenge that broad vaccines faced was antibody-dependent enhancement of viral entry (ADE, which Baric calls “vaccine-associated deleterious outcome”). In a nutshell, ADE is when vaccines instead of protecting from a virus, can actually make a subsequent infection worse because the antibodies they elicit are not only not neutralizing enough to prevent infection, but they can actually help the virus get into cells, often even immune system cells. One hypothetical way such “bad” antibodies can do this is by changing protease cleavage (e.g. by furin) via physically changing the spike protein structure due to their binding. In fact, this is something WIV and its collaborators were actively researching in late 2019, but without Ralph Baric.
Furin cleavage sites in the DEFUSE proposal
Speaking of furin cleavage sites, Dr. Baric has finally cleared up all uncertainty about what exactly the DEFUSE grant was proposing to do:
“Now, the way the furin cleavage site was built in that grant, at least in the earlier versions, some of that may have been lost as they tried to condense it to get it to fit, was that the first part was that we were fundamentally interested in why didn’t sarbecoviruses have a furin cleavage site. There had been studies done in 2010, 2011, 2012 using pseudotypes. Catherine Holmes published one in JB, there was a Chinese group that published it, where they dropped the furin cleavage site into the SARS from 2003. There was no increased infectivity, there was just a little bit more fusion between the cells. So no really big phenotype. Another example of furin cleavage sites with coronaviruses, a researcher at University of Pennsylvania knocks out the furin cleavage sites in mouse hepatitis. No change in pathogenesis for the ability of the virus to replicate. Feline infectious peritonitis virus, it’s an enteric form, it’s got a furin cleavage site, it replicates, and it got very mild infection. When the furin cleavage site is lost, it kills the cat. So it’s a flip, right? Furin cleavage site is the loss of — it’s protecting from virulent disease. So the data going into that proposal, the exact role of furin cleavage site was not clear. We were interested in it because most other coronaviruses in family had those sites. Why didn’t sarbecovirus? So the way the grant was designed was that the discovery group would look, as they did discovery, if they found one with the furin cleavage site, we would first study the pseudotypes. The second thing we would do is move it into the chimeras to see what the effect on applicants was. The third thing was we would probably build virulent viruses and study pathogenesis, and then we would knock out the furin cleavage site.”
I emphasized the part about feline coronaviruses in the quote above because not only could it explain the unusual FCS seen in SARS2, but also because it has a potential connection to ADE. But first let me elaborate on the role of furin cleavage sites in feline coronavirus pathogenicity that Dr. Baric is referring to.
Put simply, not all furin cleavage sites are created equal. Some are cleaved (i.e. cut) more efficiently, some less. The canonical pattern of an efficient cleavage site is RxRR, where R denotes arginine and ‘x’ stands for any amino acid residue (for sticklers, RxKR is also considered canonical but that’s of minor relevance to us). Now, if instead of RxRR the FCS has RxyR (where ‘x’ could be anything, while ‘y’ could be anything but R or K; for example, RRAR, RSVR, or RRSR), then it is cleaved less efficiently than RxRR, and sometimes, depending on what ‘x’ and ‘y’ actually are, it can stop getting cleaved by furin at all. But less efficient cleavage does not automatically mean a less efficient virus, as the case of feline coronaviruses illustrates.
Coronavirologists have been puzzling over feline coronaviruses for decades. Initially, they even thought that the mild enteric coronavirus and the lethal peritonitis one are two different viruses. However, step by step, they realized that it is the same virus, but one that establishes a persistent infection in a given cat and, if it then mutates in a certain way, it can become lethal. (Quick aside — new fear unlocked: how many human persistent viruses can do the same? Could shingles be caused by something similar?)
Strikingly, the mutation that kills cats is a furin cleavage site mutation that makes it less efficient! I should note that I was surprised to hear Dr. Baric describe this mutation as “knocking out” a furin cleavage site, because that is not what happens — the FCS is almost always still there, it just mutates from a more efficient, canonical RxRR form to a less efficient RxyR one.
I first learned about this aspect of feline CoVs from a Twitter thread describing the following study of feline coronaviruses that provides extremely compelling evidence that what turns a benign enteric (FECV) infection into a lethal peritonitis one (FIPV) is indeed the mutation of a highly cleavable RRSRR or RRARR furin cleavage site into something less efficient, like PRRAR:
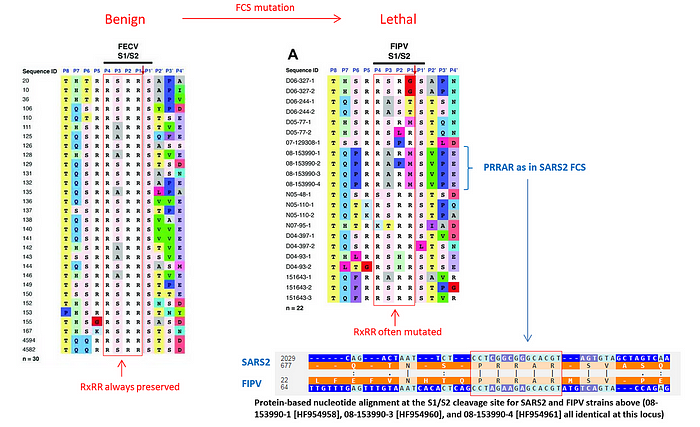
As you can see from the figure above, that study reported three different FIPV strains that had the same PRRAR furin cleavage site as in SARS2. It also mentioned an example of two cats living in the same household who were both carrying the benign enteric version of the virus but when one of the cats died, it turned out that its virus mutated its FCS from RxRR to a less efficient RxyR. This is the observation Ralph Baric was likely referring to in his testimony.
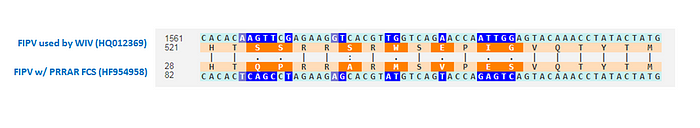
By the way, Zhengli Shi must have also known about this peculiarity of feline CoVs, and not just through conversations with Ralph Baric. In 2015, she coauthored a joint paper between WIV and Wuhan University that tested a pan-coronavirus inhibitor against several CoVs, including SARS1, MERS, and FIPV. The FIPV strain they used had the same FCS mutation as above, which turned a canonical RxRR FCS into a less efficient RRSR:
Other examples of FCS mutations changing coronavirus properties
A similar change in virulence due to an FCS mutation was observed in a human coronavirus, OC43: when it had the less efficient RRSR furin cleavage site instead of the canonical RRSRR one, it was more lethal and more neurovirulent when tested in a mouse model.
Possibly, the RRSR furin cleavage site in OC43 inspired a genetic engineer to introduce this “suboptimal” FCS, as lab leak critics call the SARS2 FCS, into another human coronavirus, NL63, which does not normally have one.
Besides feline coronaviruses, Ralph Baric also mentioned murine ones, MHV, as the inspiration behind his FCS suggestions in DEFUSE. There are two notable MHV strains when it comes to our analysis: A59 and JHM. JHM has the canonical RRARR furin cleavage site, while A59 has the less efficient RRAHR. Notably, in this case, JHM is the more lethal one and more neurotropic, while A59 is milder but does cause more severe hepatitis. The difference in their protease cleavage was suggested by University of Pennsylvania researchers as one potential factor that could explain the disparity between these strains. Notably, MHV strains generally have interesting variability in their protease cleavage sites, somewhat reminiscent of the patterns seen in feline CoVs:
Speaking of MHV, Ralph Baric and Fang Li worked on a joint paper on MHV-A59 just before the pandemic (submitted in October 2019), including investigating its protease cleavage:
It is worth noting that compared with the unliganded S-e, the proteolysis sites (at the S1/S2 region and S2’ site) do not become more exposed in the receptor-bound S-e (S4 Fig). Overall, receptor binding does not trigger dramatic structural changes in MHV S-e, which still stays in the pre-fusion conformation.
Prior research into MERS and SARS spike cleavage
The DEFUSE authors’ interest in looking for existing furin cleavage sites in SARS-like viruses and potentially engineering novel ones was preceded by similar joint work on MERS: in 2015, Ralph Baric, Shi Zhengli, Shibo Jiang and Lanying Du coauthored a paper (with Fang Li, who was a visiting researcher at WIV, as the senior and corresponding author) showing that engineering a novel FCS in a MERS-like HKU4 virus and ablating a single N-linked glycan enabled it to enter human cells.
Long before that, in 2007, Shibo Jiang, Yusen Zhou, and Lanying Du studied the role of S1/S2 cleavage in SARS1 viral entry:
We further explored if the cleavage of the S protein indeed plays a critical role in SARS-CoV infection. … here we have provided the cleavage of the S protein as another crucial target for the development of vaccines and antiviral agents. Inhibition of the cleavage of the S protein into functional S1 and S2 subunits using agents such as Ben-HCl can effectively block viral entry.
Then in 2009, Shibo Jiang, Lanying Du, and Yusen Zhou, in the paper titled “The spike protein of SARS-CoV — a target for vaccine and therapeutic development” again talked about SARS1 cleavage, and this time contrasted it with other coronaviruses cleaved by furin. In the same paper the authors mention prior work coauthored by Lanying Du on peptides that interfere with S1/S2 cleavage:
Cleavage of the S protein trimer is an important event in infection, making the potential cleavage site between S1 and S2 domains another target for development of anti-SARS-CoV agents.
Notably, two years prior to the 2015 MERS-HKU4 FCS study, Shibo Jiang coauthored a paper (for which Lanying Du is listed as the editor) that created a novel RIRR furin cleavage site (albeit not in a coronavirus) via a 12-nt insertion reminiscent of the one that has endowed SARS2 with its RRAR furin cleavage site.
Therefore when SARS2 emerged with its distinctive furin cleavage site — introduced via a very odd 12-nt insertion that also included a leading proline, just as in the MERS FCS — it is very surprising that despite all their prior work, Shibo Jiang, Lanying Du, and Zhengli Shi failed to mention the FCS in their January 2020 paper, even as they were comparing SARS2 to other SARS-like strains precisely at the S1/S2 cleavage site:
By aligning 2019-nCoV S protein sequence with those of SARS-CoV and several bat-SL-CoVs, we predicted that the cleavage site for generating S1 and S2 subunits is located at R694/S695
What is even more remarkable is that Zhengli Shi’s next major paper in early February 2020, which disclosed the RaTG13 virus, also failed to mention the novel FCS — and that paper had 29 authors. It even claimed to provide a protein sequence alignment of the S1 subunit of SARS2’s spike protein relative to RaTG13 and other SARS-like CoVs, but instead of including the full S1 protein, the authors chose to cut their alignment a few residues short of the novel furin cleavage site.
Research focus on spike cleavage, vaccines and ADE in 2019
In late 2018, Ralph Baric and Fang Li coauthored a paper on MERS and SARS spike cleavage titled “Lysosomal Proteases Are a Determinant of Coronavirus Tropism”. They were interested in how differences in protease efficiency between bats and humans shape the ability of MERS- and SARS-like viruses to jump between species. While that study focused on lysosomal proteases like cathepsin L, the authors’ clear interest in the differences between MERS and SARS spike processing is highly suggestive that investigating the role of proprotein convertases like furin in the context of SARS-like viruses was also on their mind — especially given that work on this study was done around the time that the DEFUSE proposal was being drafted, as Fang Li’s FOIA emails indicate.
In 2019, Ralph Baric’s group continued looking into proteolytic cleavage, and reported investigating two new MERS-like CoVs that both had an FCS at the S1/S2 junction. Their findings again confirmed that spike cleavage played a very important role in tissue and species tropism.
Surprisingly, Fang Li was not involved in that study, despite it being quite similar to the previous joint work he did with Ralph Baric on HKU4, MERS, and SARS. Neither was Zhengli Shi’s group, despite having coauthored the DEFUSE proposal with Ralph Baric just a year before.
In turn, Ralph Baric’s prior coauthors on the 2015 MERS-HKU4 furin cleavage site paper, Fang Li, Zhengli Shi and Lanying Du, did not include his group in their latest 2019 research, but instead were joined by Yusen Zhou to again look into the cleavage of the MERS spike while studying antibody-dependent enhancement of viral entry.
That paper was co-led by Fang Li and Lanying Du. Besides MERS, the authors also looked into how SARS1 entry into immune cells (e.g. macrophages) can be enhanced via an RBD-specific antibody that Shibo Jiang and Yusen Zhou developed previously:
To expand the above-described observations to another coronavirus, we investigated ADE of SARS-CoV entry. We previously identified a SARS-CoV RBD-specific MAb, named 33G4, which binds to the ACE2-binding region of SARS-CoV RBD (49, 50); this MAb was examined for its potential capability to mediate ADE of SARS-CoV entry (Fig. 3E). The result showed that 33G4 mediated SARS-CoV pseudovirus entry into CD32A-expressing cells but blocked SARS-CoV pseudovirus entry into ACE2-expressing cells. Therefore, both the MERS-CoV RBD-specific MAb and the SARS-CoV RBD-specific MAb can mediate the entry of the respective coronavirus into Fc receptor-expressing human cells while blocking the entry of the respective coronavirus into viral-receptor-expressing human cells.
Shibo Jiang, Yusen Zhou, and Lanying Du have been close colleagues for over a decade, having initially worked on SARS vaccines as far back as at least 2007, when both Lanying Du and Yusen Zhou still held positions with the State Key Laboratory of Pathogen and Biosecurity at the Beijing Institute of Microbiology & Epidemiology. Shibo Jiang’s focus on SARS vaccines and mAbs goes back even farther, as his 2005 and 2006 patents show.
The three researchers have also written extensively on spike-based MERS vaccines from 2013 to 2018, and coauthored joint patents on vaccines against MERS and influenza. By 2019, they have been collaborating for many years under a grant R01AI098775 titled “RBD recombinant protein-based SARS vaccine for biodefense” and have published many papers on SARS and MERS spike-based vaccines. For example, in 2017, they published a paper on a MERS vaccine that used multiple RBDs from several MERS-CoVs to generate broader immunity.
While the Primary Investigator on the above grant was Peter Hotez, in 2018, Lanying Du has gotten her own grant R01AI139092 titled “Structure-based design of coronavirus subunit vaccines”. It was a joint effort with Fang Li and Shibo Jiang:
The description of that grant indicates that the grantees were interested in novel SARS and MERS vaccines, as well as in modulating spike glycosylation, which is notable in connection with SARS2’s missing N370 glycan and the prior research of Lanying Du and Shibo Jiang, in which they removed the same glycan in a SARS1 vaccine candidate.
Under the new Lanying Du grant, in 2019, Yusen Zhou coauthored four papers: three on MERS and one on Zika vaccines:
In light of that grant and a 2017 joint paper with WIV, coauthored by Shibo Jiang, Lanying Du, and Zhengli Shi, on SARS cross-neutralization, it would be logical to assume that the frequent collaborators were also interested in broad SARS vaccines. In fact, based on a manuscript submitted to biorXiv in May 2020, Lanying Du and Shibo Jiang were indeed working on a SARS vaccine in 2019.
Fang Li too was interested in these topics, as his March 2019 talk titled “SARS and MERS coronaviruses: cross-species transmission and vaccine design” indicates:

The ADE and feline coronaviruses connection
There are notable parallels between Covid and feline infectious peritonitis. Recall that the latter is caused by persistent feline coronaviruses that mutate their furin cleavage sites to be less efficient. In light of that, and the fact that SARS2 shares the same FCS (PRRAR) as some known strains of FIPV, those parallels are even more notable. Let me quote a paper titled “Clinical and Molecular Relationships between COVID-19 and Feline Infectious Peritonitis (FIP)” about some particular similarities:
6. Molecular Similarities between the FCoV and SARS-CoV-2 Spike Proteins
…
SARS-CoV-2 is similar to FCoV-1 (and currently unique for SARS-related viruses) in that there are two identified cleavage sites (S1/S2 and S2′), with the former, the furin cleavage site or FCS, thought to be a significant factor in pandemic spread [157,158,159]. In both cases, the presence of the S1/S2 cleavage sites sets FCoV-1 and SARS-CoV-2 apart from their close family members. The importance of the cleavage activation site appears to link directly to the proteases necessary for viral infection and thus, to an additional component of tissue tropism. In type I FCoV, the transition from FECV to macrophage-tropic FIPV was first shown with amino acid substitutions at the S1/S2 cleavage site on FIP-confirmed pathology samples, which were predicted to downregulate proteolytic priming by furin-like proteases prior to S2′-mediated fusion activation [72,160,161].
The authors also mention ADE:
Antibody-dependent enhancement (ADE), the process by which viral–antibody complexes enhance infection, was of particular concern during the SARS-CoV-2 vaccine development process. FIPV has been shown to exhibit ADE in the presence of anti-FIPV antibodies [146]. This enhancement of infection appears to be specific to serotype, with passive immunization of cats against type I or type II FIPV resulting in ADE only after challenging with the same serotype for which immunization was performed [147]. As a result, ADE has been a significant challenge toward the development of FIP vaccines. In human coronavirus diseases, ADE is yet to be fully understood. In SARS-CoV, higher concentrations of anti-spike antibodies were found to have a greater neutralizing effect, whereas more dilute concentrations were suggested to contribute to ADE in vitro [148]. In SARS-CoV-2, ADE was observed in monocyte lineages but was not associated with upregulation of proinflammatory cytokines [149]. Modeling of spike protein sequences identified possible mechanisms for ADE, involving interaction with Fc receptors on monocytes and mast cells [150].
Recall that the Twitter thread that pointed out the 2013 paper on FCS mutations like RRARR -> PRRAR turning benign FECV into lethal FIPV did so in connection with the 2019 WIV paper on MERS ADE coauthored by Zhengli Shi, Yusen Zhou, Lanying Du, and Fang Li. That paper looked into how antibody binding could enhance MERS viral entry into immune cells (“Fc receptor-expressing cells”, e.g. monocytes and macrophages) by modulating spike cleavage, and referenced three papers on feline CoV ADE:
ADE has been observed for coronaviruses. Several studies have shown that sera induced by SARS-CoV spike enhance viral entry into Fc receptor-expressing cells (42,–44). Further, one study demonstrated that unlike receptor-dependent viral entry, serum-dependent SARS-CoV entry does not go through the endosome pathway (44). Additionally, it has long been known that immunization of cats with feline coronavirus spike leads to worsened future infection due to the induction of infection-enhancing antibodies (45,–47). However, detailed molecular mechanisms for ADE of coronavirus entry are still unknown. We previously discovered a monoclonal antibody (MAb) (named Mersmab1) which has strong binding affinity for MERS-CoV RBD and efficiently neutralizes MERS-CoV entry by outcompeting DPP4 (48); this discovery allowed us to comparatively study the molecular mechanisms for antibody-dependent and receptor-dependent viral entries.
The 2013 paper on the FCS mutations turning FECV into FIPV nicely summarizes that the lethality of FIPV is associated with its newfound ability to infect immune cells like monocytes and macrophages:
A long-standing hypothesis is that FIP viruses arise from internal mutation of endemic FECVs (12), which is believed to occur in approximately 1%–5% of enteric infections, resulting in the ability of the virus to infect blood monocytes and tissue macrophages. The resulting productive infection of these cells, a hallmark of FIP, enables systemic spread and results in macrophage activation, with concomitant immune-mediated events leading to death.
Recall that the authors of the 2019 MERS ADE paper led by Fang Li and Lanying Du were also interested in SARS ADE, and claimed that an antibody they previously developed can help SARS get into immune cells.
With that interest in mind, and considering their additional interest in engineering novel furin cleavage sites in SARS-like viruses, as well as searching for novel SARS-like viruses that are 10–25% different from SARS1 and likely to evade SARS1-based antibodies, which is a recipe for ADE, it is completely plausible for researchers who have just studied how MERS spike cleavage affects ADE in MERS, when they come across a MERS-like FCS in a feline CoV hypothesized to cause ADE, might want to try putting such an FCS in a SARS-like CoV to see if SARS1-based antibodies could not only fail to neutralize such a virus but could also supercharge it via similar mechanisms that they just demonstrated causes ADE in MERS.
Addressing Ralph Baric’s skepticism about FCS being engineered
In his testimony, a few hours before explaining his rationale behind suggesting to look for and engineer furin cleavage sites in SARS-like viruses in DEFUSE, Ralph Baric voiced typical arguments for why he thinks the FCS in SARS2 is unlikely to have been engineered. In short, “why use an inefficient FCS”, “why include a leading proline?”, “why create it via an insertion?”, and “why is the insertion not in frame?”:
Q: “I want to ask about the furin site a little bit. Dr. Garry, after the call, in the notes, expressed concern over — he called it a 13 nucleotide insertion that was created at the site, and said ‘I just can’t figure out how this gets accomplished in nature, but in a lab, it would be easy.’ How would you kind of refute Dr. Garry’s points there?”
A: “The sequence, you only need to insert three amino acids to make a furin cleavage site. Four amino acids went in asymmetrically. Why would anybody engineer that and do it that way, putting in an extra residue which is a proline, which puts kinks in proteins, it usually screws things up. And ultimately, that proline changed within a few — within one or two variants. So that didn’t make a lot of sense to me. But if you were going to engineer it, I guess the question would be, you don’t need to put four amino acids in, it’s easier to put three amino acids in, in the frame. And also, you’d probably want to put one in that was efficient. The sequence in SARS2 is not a very efficient cleavage site.”
Q: “So Dr. Garry was just kind of wrong?”
A: “You can make — no, I’m not saying he’s wrong. I’m just saying that means if it went in that way, then it was nefarious purposes to begin with, right? Because you’re basically trying to cover up what you did. I don’t think — I mean, when I looked at it, when it went in asymmetrically, that was more akin to recombination for me. Because recombination is not always perfect. Sometimes you have perfect recombination, but oftentimes, it’s offset and it introduces additional residue. One nucleotide or two nucleotides, depending on how it goes in, it’s sort of the random process of recombination.”
Let me address them below.
Why use an inefficient FCS?
This one is the easiest to answer, as Ralph Baric has answered it himself later in his testimony when he pointed to feline coronaviruses, where the change from an efficient to an inefficient FCS turns a benign enteric infection into a lethal one, potentially via ADE mechanisms. Thus, for researchers studying ADE and the role spike protein cleavage plays in it, picking an inefficient FCS — like PRRAR seen in multiple lethal strains of feline viruses where it causes ADE — to use for their research seems like a logical choice.
In fact, it seems that Dr. Baric himself suggested that reducing the FCS efficiency in a live virus would have been the final step in the research he proposed in DEFUSE:
The third thing was we would probably build virulent viruses and study pathogenesis, and then we would knock out the furin cleavage site.
As when speaking about the FCS mutations that turn benign feline CoV strains into lethal FIPV ones Dr. Baric referred to those mutations as “when the cleavage site is lost”, then “knocking out” an FCS could well include turning the canonical efficient FCS like RRARR into the inefficient PRRAR.
In fact, inspired by the same observations in feline and murine coronaviruses as Dr. Baric, a genetic engineer choosing to use PRRAR to “knock out” an FCS in a SARS-like strain, might well have started with RRARR as an initial efficient FCS — both feline enteric CoVs and MHV-JVH have RRARR as their FCS. For their inefficient FCS counterparts, MHV-A59 has RRAHR, while several FIPV strains have PRRAR. The latter also has a leading proline just like MERS, so a genetic engineer previously working on MERS could thus give preference to PRRAR.
Why include a leading proline?
I was actually surprised to hear this question from Dr. Baric, since he worked on MERS cleavage extensively, including creating a novel FCS in HKU4, a MERS-like bat strain. MERS, of course, also has a leading proline, as does the PRRAR furin cleavage site seen in multiple lethal feline coronavirus strains, as I just mentioned above.
Why is the insertion not in frame?
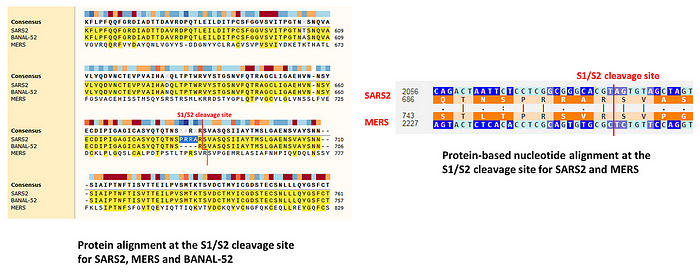
While the reading frame was obviously preserved by the PRRA insertion, the serine codon immediately preceding it is different in SARS2 than in its close relatives like BANAL-52 or RaTG13 (TCT vs. TCA):
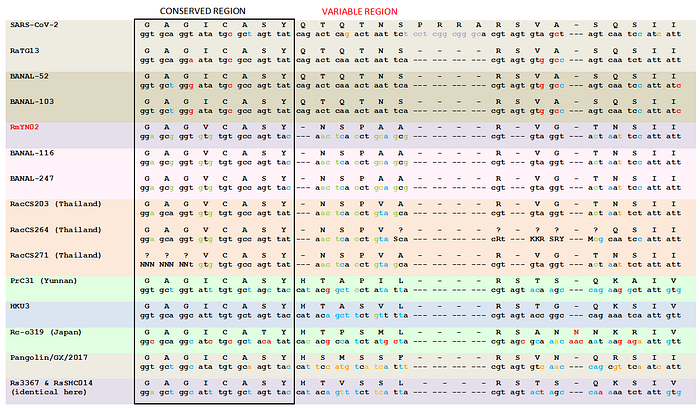
This means that at the nucleotide level, relative to those strains, the insertion in SARS2 looks as if it has split the ancestral TCA codon with the 12 inserted nucleotides.
However, there are at least two other alternatives. One is that the direct progenitor of SARS2 prior to the insertion already had TCT for its serine, and the insertion of 12 nucleotides was fully in frame. Another possibility is that the direct progenitor of SARS2 prior to the insertion had TCA for its serine just like BANAL-52 or RaTG13, then the in-frame insertion happened, and then at some point between that and getting noticed in the first human patients, the TCA codon mutated to TCT.
Both scenarios are plausible, as SARS2 is hundreds of mutations away from its closest relatives like BANAL-52 or RaTG13, and several of those mutations are present in close proximity to the TCT codon in question, as can be seen in the figure above. In fact, the S1/S2 junction is generally a mutational hotspot, and under a lab leak scenario, the engineered virus would subsequently be subjected to testing in cells and/or animals, acquiring additional mutations before spilling over to humans.
Why use an insertion to create the FCS?
In line with the scenario where researchers studying the role of spike protein cleavage in MERS ADE were now looking to extend their research to SARS-like strains, the desire to equip a SARS2 progenitor with a MERS-like FCS can also explain why a genetic engineer might have chosen to do so via an insertion rather than mutation of existing nucleotides: the MERS spike protein is four residues longer at the S1/S2 junction than SARS-like viruses, and an insertion like PRRA removes this imbalance:
To see what impact the insertion has on the 3D structure of the protein, I aligned 3D structures of the MERS spike with two versions of the SARS2 spike: one where the FCS would have been created without an insertion (i.e. via mutation of existing residues) and another one as found in SARS2 normally (i.e. where the FCS arose via the PRRA insertion). It turned out, thanks to the PRRA insertion, the S1/S2 loop in SARS2 has its cleavage point ~3x closer to the MERS one than if the FCS in SARS2 was engineered without an insertion (i.e. via mutation of existing residues).
Here is a snapshot of the three spikes and the distances between the S1/S2 junctions relative to MERS (orange) between SARS2 (green) and a SARS2 mutant that has its FCS arise without an insertion (pink):
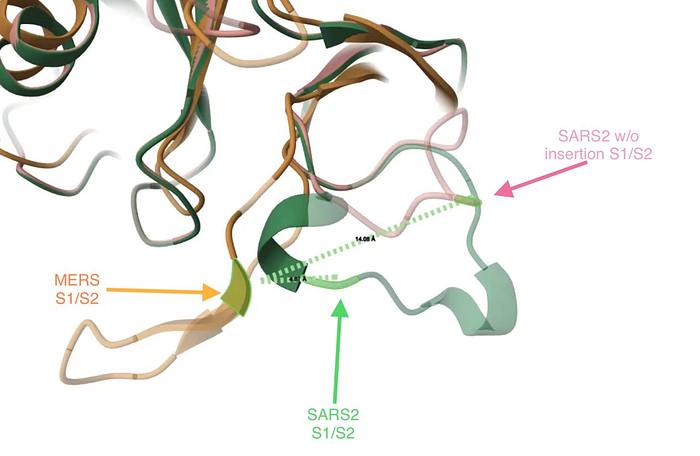
For a detailed explanation, see my 3D protein modeling video in this Twitter thread:
So if a genetic engineer was interested in creating a MERS-like FCS in a SARS2 progenitor, doing it via an insertion makes sense, especially in connection with studying ADE in SARS-like viruses.
In line with this hypothesis, SARS2 has two interesting unique mutations in its receptor binding domain (RBD) not seen in any other known SARS-like strains: R346 and A372. The first one is reminiscent of a SARS1 escape mutation from the S109.8 antibody, while the second removes a highly conserved N-linked glycan previously implicated in DC-SIGN binding (a receptor on immune cells involved in ADE) and ablated in a SARS1 vaccine candidate in a 2013 paper coauthored by Lanying Du and Shibo Jiang on RBD-based SARS1 vaccines. I described both of those unique SARS2 mutations in detail in my previous article:
In that article, I also review how in line with Ralph Baric’s explanation about the necessity to broaden the panel of viruses to test novel vaccines and therapies against, EcoHealth and WIV updated their virus hunting criteria to now look for SARS-like viruses that were 10–25% different from SARS1 in their spike but could still enter human cells. To accomplish that, they proposed sampling 5,000 bats in order to “almost fully characterize the expected natural diversity of SARSr- and other β-CoVs in the region”.
SARS2 fits those criteria like a glove: its spike is 24% different from that of SARS1, and yet it binds to the human ACE2 receptor even better than SARS1. Moreover, since SARS2 or its BANAL-like progenitor would have been able to evade SARS1-based vaccines and antibodies, such strains would have been prioritized and likely turned into full-genome synthetic backbones, as the joint EcoHealth/WIV grant renewal application suggested.
Pangolin coronaviruses in 2019
All of the lab leak puzzle pieces relating to relevant research done in 2019 in Wuhan are in place. The only missing piece is a SARS2 progenitor strain at WIV. But there are other known puzzle pieces that strongly suggest that, given the interests of WIV and its vaccine designing collaborators, they could have been actively searching for a BANAL-like progenitor of SARS2 in 2019. Those puzzle pieces are two SARS-like coronaviruses found in pangolin samples that were being actively researched in 2019. One such coronavirus is now known as GX/2017, and the other as MP789 or GD/2019.
GD/2019 is the most interesting one because it has a 97% identical receptor binding domain (RBD) to SARS2. It was found in tissue samples of confiscated contraband pangolins, whose potential infection by SARS-like viruses was first reported by the Guangdong Institute of Applied Biological Resources (GIABR), a close collaborator of WIV.
GIABR submitted their paper in September 2019. In it they described sequencing tissue samples collected earlier in 2019 from confiscated pangolins, and reported finding coronavirus genome fragments that matched to a number of SARS-like viruses. Most notably, in the “lung08” sample they reported a 500-nt fragment that aligned to the spike of the HKU3 virus, with a 82.6% identity. HKU3 was listed as one of EcoHealth/WIV’s viruses of interest in the DEFUSE proposal and the November 2018 NIH grant renewal application.
After the outbreak, metagenomic analysis of the sequencing reads from the same “lung08” sample showed that it contained reads matching the SARS2 receptor binding motif (RBM, the part of the RBD that comes into contact with the ACE2 receptor), and pooled reads from lung07, lung08, and lung09 samples would later produce a near-complete genome of the MP789 virus that shares 97% identity with SARS2’s RBD but, more importantly, has a near-identical RBM with SARS2: they share 77 out of 78 residues, including all six residues critical for efficient binding to ACE2.
The senior author of that 2019 GIABR paper on pangolin CoVs, Jinping Chen, collaborated with WIV and EcoHealth before — for example, on a 2014 paper sampling multiple bat species at the Xishuangbanna Tropical Botanical Garden in Yunnan, near the border with Laos, within a few kilometers of where a very notable SARS-like virus, RmYN02, was found in 2019. So it is quite possible that WIV was aware of GIABR’s findings regarding identifying a number of SARS-like viruses in pangolin samples. In that case WIV would have likely had access to those samples and/or sequencing data in 2019.
The GIABR analysis of its pangolin samples could have been spurred on by the earlier discovery of GX/2017 by Yigang Tong of the “State Key Laboratory of Pathogen and Biosecurity” at the Beijing Institute of Microbiology and Epidemiology — the same lab as Yusen Zhou’s. In fact, in their February 2020 paper, Dr. Tong’s group claims to have isolated GX/2017 long before the Covid outbreak. On May 1, 2024, Peter Daszak testified that he was familiar with Yigang Tong’s discovery even before publication, as they have discussed this during their in-person meeting:
Dr. Tong did collaborate with WIV and EcoHealth previously, e.g. in 2018 on a SADS-CoV paper titled “Fatal swine acute diarrhoea syndrome caused by an HKU2-related coronavirus of bat origin”. Libiao Zhang of GIABR, a longtime bat sampling partner of WIV, is also on that paper. But I am more intrigued about the collaboration between Yigang Tong and Yusen Zhou, as they both hail from the same lab. For example, in 2016 they published a joint paper.
What is even more intriguing is that Yigang Tong has filed his own patent for a SARS2 vaccine, and he did so on January 22, 2020 — a whole month prior to Yusen Zhou’s patent! That’s some speed: the SARS2 genome was only made public on January 10, and Wuhan didn’t even go into lockdown until January 23, and yet, by January 20 — judging by the text of the patent — Dr. Tong has already finalized his application.
If in 2019 Peter Dazsak was aware of a new SARS-like CoV found in pangolin samples by Yigang Tong, then Zhengli Shi, Yusen Zhou, Lanying Du, Shibo Jiang, and Fang Li must have been aware of it as well. They were also likely aware of the GIABR discovery. If they had access to even partial spike sequences of those viruses or raw sequencing data, WIV and its collaborators could have searched for RBD fragments of the spike that fit their “10–25% different from SARS1" criterion.
In that case, they would have seen that GX/2017 and GD/2019 RBDs not only fit that bill but they also show high similarity to another interesting strain that WIV sequenced in 2018, RaTG13 (or Ra4991, as it was known at the time). All of these strains are also antigenically different enough from SARS1 to escape SARS1-based antibodies, even more so than WIV1 and SHC014:
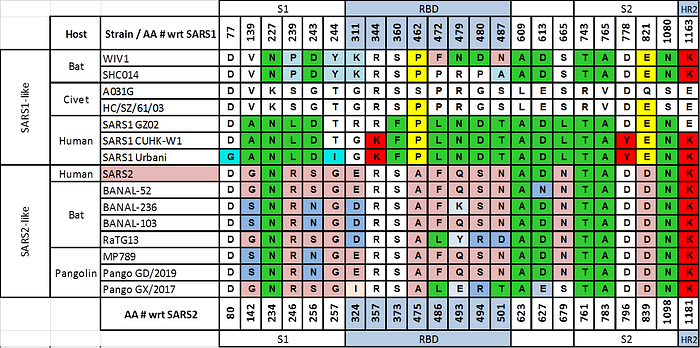
Moreover, in terms of the ability to bind to the human ACE2 receptor, computer modeling would have shown a progressive increase from RaTG13 to GX/2017 and finally to GD/2019.
That would have likely prompted a further search for viruses that shared the same RBD as GD/2019 — most likely via deep sequencing of pangolin samples and/or bat samples. Such search could well have produced a BANAL-52-like SARS2 progenitor which would then have been subjected to the kind of research described in DEFUSE and EcoHealth grants, including engineering a novel FCS and studying its ability to evade SARS1-based antibodies, as well as the propensity of those antibodies to cause ADE.
In fact, according to the declassified US Intelligence (ODNI) report on Covid origins, in 2019, WIV was analyzing at least some pangolin samples:
• Since 2019, some WIV researchers analyzed pangolin samples to better understand disease outbreaks in these animals.
…
• As of January 2019, WIV researchers performed SARS-like coronavirus experiments in BSL-2 laboratories, despite acknowledgements going back to 2017 of these virus’ ability to directly infect humans through their spike protein and early 2019 warnings of the danger of this practice. Separately, the WIV’s plan to conduct analysis of potential epidemic viruses from pangolin samples in fall 2019, suggests the researchers sought to isolate live viruses.
Thus, judging by the puzzle pieces around the only missing piece of a direct SARS2 progenitor, it seems that WIV could also have been looking for it back in 2019.
Addressing Ralph Baric’s arguments for natural origin
While Dr. Baric does not rule out a lab leak, he leans strongly towards a natural origin. Before I address his reasons for it, I want to highlight that despite his views, Dr. Baric does not think the Huanan market was the source of the outbreak, and he does not subscribe to the two-spillover hypothesis:
Q: “The 177 official WHO China corona reported cases, if you put the molecular clock to mid-October, then all of the activities around that — the market in Wuhan is actually two months or so?”
A: “It’s a major problem with that Wuhan study — that market study, yes.”
Q: “Can you just elaborate on that a little bit? I don’t have the expertise.”
A: “Okay, so keep it in context. The context is, what do you have data for? And the only thing we have really solid data for is that the market was the site of amplification in late December, January. That’s still two months from the origin date, based on a molecular clock, which means it was circulating somewhere before it got there. And the question is, where was it?”
Q: “To that point, I guess without getting too far away from our next set of questions, how hard — you’re talking about several hundred, if not several thousand human cases by the time you’re getting into January — early January, late December?”
A: “Remember that 90 percent of those cases are asymptomatic.”
Q: “Right.”
A: “85, 90 percent. So imagine trying to chase a transmission cycle. Early cases are almost impossible, because most — many asymptomatics are in the middle of it. So now you have a case here and a case here, but they’re actually truly linked by someone in the middle.”
Q: “Who just walked around with it.”
A: “Yeah. And you can’t unravel that transmission cycle until you do deep sequencing on both of them. And then you look for SNPs, and you can say, this patient is linked to this patient. It had to go through somebody else because there’s another marker. So all that — so it’s a fundamental problem with the papers that are reported to prove — they write it too strong, I think, but they’re very passionate about their data. And to be fair to them, it is the best data that’s out there, that they can’t — they don’t have the early cases. What they have, they have the cluster in the market and they have two SNPs, which they argue are indicative of two different zoonotic introductions, which other people argue with. It’s one nucleotide that’s making that call, so it — actually claimed there were two independent introductions.”
Q: “And they had some — “
A: “It’s a stretch. It’s a stretch. There are a lot of virologists that look at that data and go, mmm.”
Q: “Because it looks like, as I understand those two differences between the two lineages, it’s one looks marginally more like an ancestral bat virus?”
A: “Yes.”
Q: “And one looks a little more humanized?”
A: “At one nucleotide level. And they don’t know what the ancestral bat virus really was.”
Q: “Sure.”
A: “So from my perspective, clearly, the open market was a conduit for expansion of the disease. Is that where it started? I don’t think so.”
Q: “Keeping in mind the Chinese government’s ability to cover things up, is it at all worrisome to you or notable to you that we don’t have a second market or a third market or additional lineages coming out of nearby cities, like we saw with SARS1, where you had sort of a wave of spillover into the human population?”
A: “Remember that the Chinese Health Minister, I think on like the 24th of January, said community spread was rampant and asymptomatic spread was rampant. And they quarantined.”
Q: “A lot of people.”
A: “Within a few days of that, they quarantined 65 million. They came in and cleaned the market in Wuhan on, like, the 30th of December. What I don’t know is whether they went to every other market in Wuhan and other surrounding large metropolitan areas, or when they found them, they just wiped out — they cleaned those out. I don‘t think — I don’t have any information on it. I don’t know if you have any information on it.”
Q: “Not that we’ve seen.”
Incidentally, mid- or late-October spillover is well within range of the purported hospitalization in early November of the three members of the Zhengli Shi’s lab, reported as potential initial patients. Here is what Dr. Baric said about this:
Q: “The last major point that ODNI states is that there were Wuhan Institute researchers that were ill in the fall of 2019. The illness doesn’t necessarily support or refute either hypothesis or prove that it came from a lab. Did you have any awareness of any Wuhan Institute researchers being sick in the fall of 2019?”
A: “I’ve heard this report, but I’m not — and I’ve heard that they’ve been named, but I haven’t actually seen any of the data that supports that. So I don’t know how authentic it is. I mean, there’s, what, 5, 600 people who work in the Wuhan Institute of Virology. I don’t know the full number, but — and there was flu going on at the time, so it wouldn’t surprise me if they got sick. And I believe they — if they’re just getting physicals, they go to the hospital. So that’s their medical care system. So looking at it from that point of view, that doesn’t tell me anything.”
Q: “Okay.”
A: “I will also note one other thing. If you look at the molecular clock of the virus, it emerged in the middle of October, late October, not the middle or end of November. So people who say that those were the first cases, no chance. There were five or six transmission cycles at least before they would have been infected.”
The WIV staffers in question are actually reported to have gone to the hospital in early November:
Ben Hu and his colleagues Yu Ping and Yan Zhu were identified in media reports in June 2023 as the three workers who fell sick. All were from Shi Zhengli’s laboratory at the Wuhan Institute of Virology. Intelligence has suggested they were hospitalised in the first and second week of November — well before a cluster of cases in December were traced back to the Wuhan wet market.
So the timing of their illness does not contradict Dr. Baric’s views on the timing of SARS2’s spillover.
Also, if the Covid outbreak didn’t start at the Huanan market in Wuhan, where else could it start naturally, and get to Wuhan without infecting anyone else along the way? The nearest bat habitat is over a thousand miles away. Here is Dr. Baric’s explanation for why he feels zoonosis is much more likely than a lab leak:
Q: “So for a little bit of historical context, for zoonotic jumps with coronaviruses or even other viruses in general, could you just talk a little bit about how zoonotic jumps would happen or have happened?”
A: “In the context of coronaviruses?”
Q: “Or any other viruses, if that makes it easier for you to talk about.”
A: “Well, the first thing that has to happen is that human populations have to come into close contact with animals that encode these viruses. So that’s obviously the first thing. So there are, like, people in the extractive industry who may be loggers or hunters or, you know, gathers or collects bushmeat, those kind of people are the most likely to come into contact with zoonotic viruses and become infected. Now, the vast majority of contacts where zoonotic viruses actually are introduced into a human being, most of those don’t progress. The recent data with coronaviruses, for example, that was published in Southeast Asia argues that there’s somewhere between 50 to 60,000 exposures where people working with bats come in contact with bat coronaviruses, and actually seroconvert. That means they get infected, probably had very mild disease and recovered, 50,000. So if you think about how many — well, let’s put it in the context of coronaviruses. So 2002, SARS emerged; 2019, SARS2 emerged. That’s 17 years times 50,000 exposures a year, it’s actually a little higher. So about a million exposures between human disease outbreaks. So the vast majority of exposures are self-contained and do not transmit to another person, and then do not establish or colonize the new population. But this is occurring all the time. And so when you get to origins, for example, and you ask the question, what’s more likely, is it a lab leak or is it natural processes? You’re looking at one in a million, a million exposures occurring over 17 years versus what happens in a laboratory setting. No chance it’s even close. And the diversity in nature, hundreds of millions of times more diverse than what was in the Wuhan Institute of Virology. So that gradient is huge. And if you consider that, it’s more likely to be a natural event than it is to come out of the laboratory. The data — that’s what the data screams.”
…
Q: “And when we’re looking at the SARS-CoV-2 or COVID-19 pandemic, it sounds like you feel strongly that it was a zoonotic or natural origin. But would you say that it’s not settled yet what the origin of the COVID-19 pandemic was?”
A: “Again, I have at different times speculated on three possibilities. The first is natural origin. The second is accidental escape from the laboratory setting, which can also include collection, which you can ask about if you’d like more details on that. And then the third would be the possibility of engineering. There is no hard evidence to support engineering. Initially, for example, the receptor binding domain was argued to be completely unique and perfectly positioned, perfectly designed to bind the human ACE2 receptor. Well, no, there are virtually identical strains in bat strains that are found in nature. So it’s not been engineered. In addition, that spike has undergone successive sets of — the RBD has gone successive adaptive changes that increase bind infinity for the ACE2 over a thousand fold. It is not perfectly designed. It’s just like the origin SARS1, which underwent specific changes that enhanced its transmissibility as it was spreading. The exact same process. So the RBD is out. The second idea that it was engineered, there was a very bad bioinformatic paper, for example, that said — it came from the HIV — which was total nonsense. The better argument was that there might be a super antigen site, but there was a paper that was just published that said, no, there’s no super antigen site. So, in essence, the scientific process says, okay, if this is the hypothesis, let’s do experiments to see if we can disprove it. If we can’t disprove it, then it’s likely. So far there’s no backbone genome that’s close enough to have been engineered in the SARS2. Most of the components that were originally argued as being engineered failed. The only one that’s left is the furin cleavage site, which has multiple explanations. So that leaves two possibilities. The first is escape from the laboratory. And you can’t rule that out, because they do work at BSL-2. You just can’t. But for the reasons I talked about earlier, just on the frequency and the exposure level in nature versus lab, it’s massively — what’s that called, massive — the scales are massively weighted to natural origins, yes, sorry.”
Q: “Sure. And taking out bioengineered, I think there’s much consensus that that is not what we’re looking at here. But with the lab leak and zoonotic, there would be possibilities for it to be somewhat more of a combination of the two. I’m thinking about, specifically, you said researchers go out and collect samples, they bring them back to the lab. Maybe they do no manipulation on it, so it’s just whatever they collected out in nature. Something happens, there’s a lab accident, and somebody is exposed to a virus and gets infected. While I understand this would be very rare, that would sort of be a combo of a lab accident with a natural virus, correct?”
A: “Yes, and still be a natural virus that inadvertently escaped the laboratory, because biosafety practices weren’t sufficiently robust. Now, when you think about collection, at least the group at EcoHealth and the groups that they collaborate with, again, I haven’t been in the cave with them, but the pictures that I have seen is they’re fully dressed in Tyvek suits and with all the protective gear. So, in essence, they are collecting — in essence, in laboratory appropriate conditions, and then bringing the samples back. Their weakness is trying to culture the viruses at BSL-2. It’s just the chance of an accident is increased under BSL-2 conditions, as compared to BSL-3.”
In a nutshell, Dr. Baric feels that a natural origin is much more likely because he believes there are 50–60 thousand people quietly getting infected with SARS-like viruses in Southeast Asia every year. Where did he get this number? Turns out, from Peter Daszak and Zhengli Shi.
I won’t go too deep into their analysis, although I do feel that their methodology is flawed. For example, the key metric of their entire paper, how often contact with bats results in a detectable SARS-like infection, is based on poorly substantiated data — just a single observation of seropositivity out of 199 tested people who had contact with bats. Moreover, there is some weirdness with that number, tucked away in Supplementary Table 4:
The reference 19 used to substantiate it points to a 2019 paper by the same authors which previously reported:
Serological testing of serum samples from 1,497 local residents revealed that 9 individuals (0.6%) in four study sites were positive for bat coronaviruses, indicating exposure at some point in their life to bat-borne SARSr-CoVs (n=7, Yunnan), HKU10-CoV (n=2, Guangxi), or other coronaviruses that are phylogenetically closely related to these. All individuals who tested positive (male=6, female=3) were over 45 years old, and most (n=8) were making a living from crop production.
How the authors extracted a subset of 199 tests of the people who had contacts with bats from the above data is unclear. The only plausible explanation is a subset of 201 people who reported having bats in their house, of whom 1 tested positive for some coronavirus (as indicated by the red number 1 in their table):
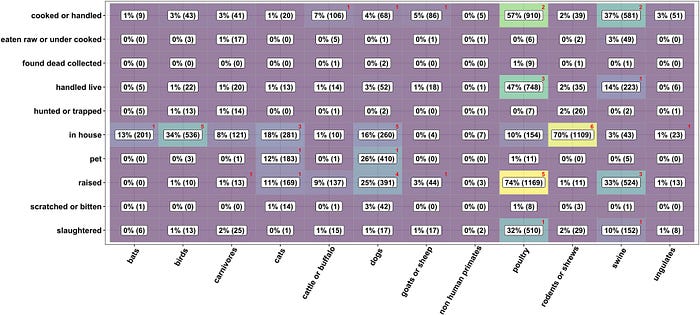
However, the 2022 paper citing those 2019 data has two instances of positive tests — one for a SARS-like virus and another one for HKU10-CoV, while the 2019 paper does not report any co-infections, and both HKU10-CoV positive tests are reported to have been from Guangxi, while all seven SARS-like CoV positive ones from Yunnan.
But the biggest observation from the 2019 data is that 8 of the 9 people (out of 1,497) testing positive for exposure to SARS-like or HKU10-CoVs reported no contact with bats! This undermines the 2022 paper’s entire thesis that it is contact with bats that is the primary source of human exposure to these viruses. Clearly, that wasn’t the case in their 2019 paper.
Curiously, before the Covid outbreak, Peter Daszak and Zhengli Shi were claiming the risk of SARS-like spillover was quite low. In their 2018 paper titled “Serological Evidence of Bat SARS-Related Coronavirus Infection in Humans, China” they reported that only 6 of the 218 villagers living within 1–6 km of the caves WIV sampled in (including Shitou cave where the SHC014 virus was found) were seropositive for SARS-like viruses. Ironically, 240 Wuhan residents were used as control.
In a wider follow-up study published in September 2019, just before the outbreak, Peter Daszak and Zhengli Shi reported even lower seropositivity prevalence — 0.6% (9 out of 1,497) that I already quoted above.
However, after the outbreak happened, Peter Daszak was pointing to their earlier, higher seropositivity prevalence (6 out of 218 villagers, who were originally called a “high risk group of residents living in close proximity to bat colonies” in the 2018 paper), and extrapolating their findings to all of SE Asia, arriving at “1–7 million people per year exposed to bat origin SARS-related CoVs”:
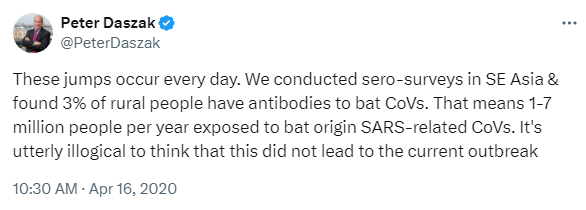
Ultimately, “1–7 million people per year exposed to” evolved into “66,280 people … infected with SARSr-CoVs annually in Southeast Asia”, and that became the pro-zoonosis talking point that Ralph Baric based his assessment of the likelihood of Covid’s natural origin on.
It is interesting how the authors also expanded their focus from China to all of Southeast Asia. This is another problem with using their 2022 paper to infer Covid origin: out of the 499 million people they included in their calculation, less than 10% live in China, while the majority live thousands of kilometers away from Wuhan — e.g. 151 million live on the island of Java alone. Thus, the relevance of such distant bat exposure to an outbreak in Wuhan is almost as moot as it would have been if the Covid outbreak happened near Ralph Baric’s lab in North Carolina — just because such a virus could have spilled over naturally thousands of kilometers away doesn’t explain why it actually spilled over in an urban center near a lab studying viruses just like it, 1500 km away from its bat habitat.
Coming back to Ralph Baric’s argument, it is also odd that he brackets the timeline of SARS2 emergence by 17 years since the emergence of SARS1, because SARS2 didn’t have just 17 years to emerge naturally to cause a Covid-like pandemic, it had decades if not centuries. Yet, it emerged only a year after WIV and EcoHealth, inspired by Baric, started to look for SARS-like viruses just like it: with a spike 10–25% different from SARS1 that is still able to infect human cells while evading SARS1-based antibodies.
Moreover, miraculously, the virus that WIV planned to hunt for in remote bat caves in Yunnan, just happened to emerge on its doorstep. Should I even mention that the virus also had a unique feature that WIV and its DEFUSE coauthors wanted to look for and engineer, a furin cleavage site? Which no other known SARS-like virus has. All of these incredible coincidences, as well as their timing synchronized with a shift in WIV’s research interests should skew probabilities much more towards a lab leak than zoonosis.
The lab leak case
Let me outline a potential lab leak scenario based on all of the above. In 2018 and 2019, US and Chinese coronavirologists were engaged in intense research focused on vaccine development and ADE in the context of developing broad-spectrum vaccines for SARS-like and MERS-like viruses. Ralph Baric was working on a US-based vaccine platform, while Lanying Du, Yusen Zhou, Fang Li, and Shibo Jiang were doing a lot of vaccine research in China.
Despite previously working closely with Ralph Baric and EcoHealth, by 2019, Lanying Du, Fang Li, and Shibo Jiang have significantly decreased their collaboration. Conversely, their joint work with WIV increased, and some research plans outlined in DEFUSE were likely now being done by WIV in collaboration with those researchers rather than Ralph Baric or EcoHealth. On the vaccine research front, WIV was now working with Lanying Du’s husband, Yusen Zhou.
By 2019, WIV held all the cards, as by then they had humanized mouse models and genetic engineering expertise, and, most importantly, they had access to bat caves, while their foreign collaborators were at their mercy when it came to finding novel “interesting” viruses, as Dr. Baric testified:
A: “It would have been going in — if year 6 was around 2019 or 2020, that’s when I would have been a part of [the EcoHealth/WIV grant]. And my role was to study a couple of the viruses that the Wuhan Institute of Virology found that they were willing to share with me. So I always viewed that as not number one or number two on the list, maybe number five or number six on the list.”
Q: “I understand, I think I understand what you’re saying. But when you say not one or two on the list, but number five on the list, is that as far as they are giving you the fifth most interesting virus that they had found?”
A: “Well, to be fair to them, they did the discovery work and they’re going to choose the priority of what they want to work on first. And so I’m not going to get the dregs, that would be an unfair characterization, but I’m not going to get number one. I’m going to get somewhere down the list, which is okay, and I understand that process. Hopefully, it would be something that they felt would be interesting as well.”
Needing to expand their diverse panel of viruses to evaluate the effectiveness of new broad vaccines, WIV and its collaborators kept looking for novel viruses. In particular, for SARS vaccines, they were interested in novel SARS-like viruses, differing from SARS1 by 10–25% in its spike protein. They were also interested in strains that could escape SARS1-based antibodies, which at the same time could lead to ADE.
Inspired by Ralph Baric’s ideas about furin cleavage sites in SARS-like viruses, as well as his joint work with Fang Li on SARS and MERS spike cleavage, WIV and its collaborators were also likely looking to engineer furin cleavage sites in novel SARS-like strains that fit their search criteria. Based on their interest in vaccines and ADE, extending their investigation of MERS ADE to novel SARS-like CoVs that could escape SARS1-based antibodies seemed like a logical next step, as did using an FCS from feline coronavirus strains known to exhibit ADE, especially since that FCS was similar to the one in MERS.
So when in late 2019 SARS2 emerges against this backdrop in Wuhan — with its spike 24% different from SARS1 and yet extremely adept at binding the human ACE2 receptor, and with a MERS-like furin cleavage site identical to the one found in several lethal feline coronaviruses — it is much more plausible that this virus materialized in Wuhan through the research that was being conducted there, rather than by a series of incredible coincidences in nature.
